锌配合物催化二氧化碳和环氧丙烷环加成反应机理研究
2018-04-02杨良艳冯华杰李志伟
尹 燕,杨良艳,冯华杰,李志伟
(1.肇庆学院 环境与化学工程学院,广东 肇庆 526061;2.海南师范大学 化学与化工学院,海南 海口 571158)
0 引言
固定活化利用CO2一直是研究的热点,能提供丰富碳源,减缓温室效应,符合绿色化学和可持续发展要求[1-5].用CO2可以制备多种重要化合物,如CH3OH、环碳酸酯等,可用催化剂种类很多,如金属、金属配合物、离子液体等,各种催化剂均可取得良好催化效果[6-12].锌配合物在CO2固定活化利用中适用范围广、催化活性高、价廉、反应选择性高、可循环使用.尽管进行了大量实验研究,但对于金属催化CO2活化的反应机理还没有完全明确,一定程度上减缓了CO2固定活化利用研究进程.
He[5]等研究了Zn(OPO)2/TBAI(Zn(OPO)2结构如图1,TBAI:四丁基碘化铵)催化体系对环氧化合物和CO2反应的实验结果,该催化体系在此类反应中具有极好稳定性和高效性,如图1所示.研究通过密度泛函理论对其进行计算,为进一步明确反应机理从而更好地设计高效、稳定、条件温和的催化剂提供理论支持[13,14].
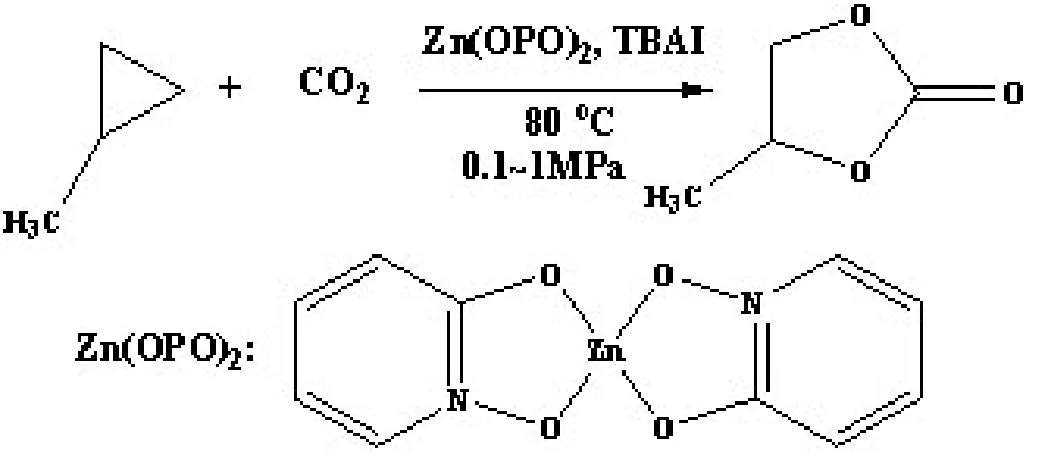
图1 Zn(OPO)2/TBAI催化二氧化碳和环氧化合物的环加成反应
1 计算方法
采用密度泛函理论的B3LYP方法,I和Zn原子采用LANL2DZ ECP基组,其他原子使用6-31G(d)基组,对反应物、中间体、过渡态和产物进行结构优化和频率计算,确保中间体没有虚频,而过渡态有且只有1个虚频.对过渡态结构利用内禀反应坐标(IRC)方法对反应路径进行验证,确保所有过渡态均可通过势能面连接相应的反应物和产物.此外,利用前线分子轨道理论分析反应的成键情况.所用的能量均采用校正后的吉布斯自由能,所有计算都在Gaussian 09[15]程序包中完成.
2 结果与讨论
2.1 反应过程机理
在催化反应过程中,反应可能经过以下步骤完成.首先,C3H6O与Zn(OPO)2中作为路易斯酸性中心的Zn原子配位得到σ键配合物成为亲电质点,接着作为亲核体的I-进攻C3H6O上的C原子,得到开环产物;然后,开环产物作为亲核试剂进攻CO2的C原子,得到金属碳酸盐;最后,金属碳酸盐发生分子内的闭环反应,还原催化剂得到环碳酸酯,可能机理如图2所示.根据图2的路径,用密度泛函理论进行计算.
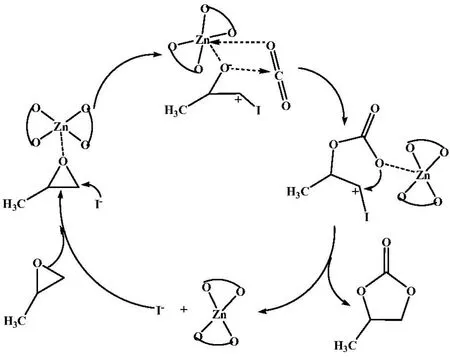
图2 Zn(OPO)2催化CO2和环氧丙烷的环加成反应机理
机理假设考虑了2种反应途径.途径A中,I-进攻方向与C3H6O上的甲基在异侧.途径B中,I-进攻方向与C3H6O上的甲基在同侧.途径A和B的相对吉布斯自由能如图3所示.
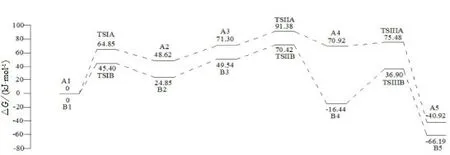
图3 途径A和B的相对吉布斯自由能图
图4给出了途径A涉及的反应物、中间体、过渡态和产物的分子构型.首先,作为路易斯酸性中心的Zn原子与C3H6O配位形成σ键配合物A1,从而活化C3H6O上的C2-O键促进其开环,接着亲核体I-从甲基的异侧进攻C3H6O的C2-O键,经过过渡态TSIA,形成中间体A2,此过程需克服64.85 kJ·mol-1的能垒.从图4中可见,A1中C2-O键的键长(0.145969 nm)比过渡态TSIA中的键长(0.18682 nm)短0.040851 nm,A2中C2-I键的键长(0.230760 nm)比过渡态TSIA中的键长(0.281668 nm)短0.050908 nm.另外,TSIA有且只有1个虚频(-325.65 cm-1),虚频的振动方式也说明I-的亲核进攻伴随着C2-O键的断裂与C2-I键的形成,即TSIA是连接A1和A2的正确的过渡态.随后,CO2与A2加合形成A3,经过过渡态TSIIA(-135.37 cm-1),CO2插入Zn-O键形成A4,此过程需克服20.08 kJ·mol-1的能垒,同时导致了Zn-O1键、C-O键的形成与Zn-O键的断裂.TSIIA虚频的振动方式证明其为连接A3与A4的正确过渡态.最后,A4经历分子内的闭环反应得到最终产物A5,此过程需经历过渡态TSIIIA,虚频为-367.93 cm-1,其振动也是开环和闭环的方向,伴随着C2-I键与Zn-O1键的断裂而C2-O2键的形成,该反应步骤的能垒为4.56 kJ·mol-1.所以,经过3个步骤,生成了最终产物环碳酸酯,整个反应的决速步是CO2的插入固定,整个反应放出能量40.92 kJ·mol-1.
因为途径B与途径A基本相似,差别在于I-进攻C原子的方向不同.反应过程中反应物、过渡态、中间体、产物的结构如图5所示,而相应吉布斯自由能如图2所示.途径B中3个反应所需克服的能垒分别为45.40、20.88和53.35 kJ·mol-1.与途径A相似的是,途径B中CO2的插入固定也是决速步,ΔG值为70.42 kJ·mol-1,同时途径B也是放热反应.与途径A不同的是,途径B的3个反应步骤的能垒相对于途径A分别低了19.45、20.96和38.58 kJ·mol-1,表明途径B比A所需能量更低,是更有利的反应途径.这可能是因为途径A中,I-在甲基的同侧进攻C2原子时,C1上甲基供电子效应更大造成的.

图4 途径A的相关分子构型图
2.2 前线分子轨道理论分析
为进一步解释目标反应机理,运用前线分子轨道理论对化合物A1、B1和CO2进行分析.
由图6可知化合物A1的LUMO轨道与CO2的HOMO轨道能量相差较大,为867.99 kJ·mol-1;而化合物A1的HOMO轨道与CO2的LUMO轨道能量相差较小,为473.48 kJ·mol-1,可见反应由A1提供电子,CO2提供空轨道.CO2的HOMO轨道是由O1和O2原子的p轨道组成,其LUMO轨道则是由C原子上较大的p轨道成分组成;而化合物A1的HOMO轨道由O原子上的p轨道组成,其LUMO轨道则由C2原子上较大的p轨道成分组成.所以,轨道成分有利于C-O键的形成,其次才是有利于C2-O2键的形成,这很好地符合了途径A的反应机理过程.
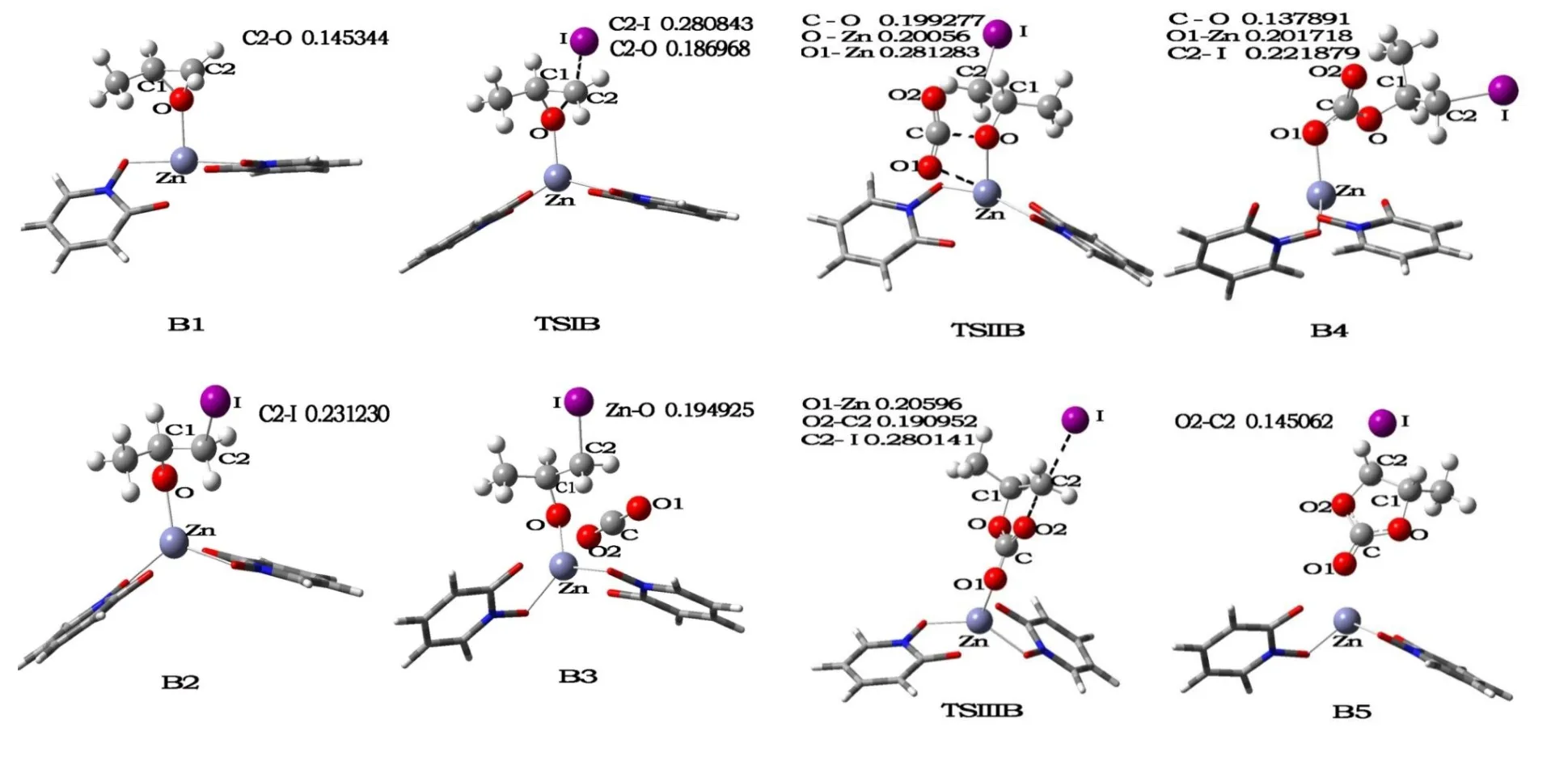
图5 途径B的相关分子构型图

图6 前线分子轨道的对称性和能量
对化合物B1,发现当I原子与甲基在同侧时,相比化合物A1,延长了CO2的O原子和化合物B1的甲基C原子之间的距离,这导致了化合物B1的HOMO轨道和CO2的HOMO轨道之间的排斥作用变弱,这更有利于C-O键和C2-O2键的形成,所以很好地解释了途径B优先于途径A的原因.因此,前线分子轨道理论分析进一步验证了Zn(OPO)2催化CO2和C3H6O的环加成反应机理.
3 结论
利用密度泛函理论的B3LYP方法,对Zn(OPO)2配合物催化CO2和C3H6O的环加成反应机理进行了理论研究.结果表明,反应过程中,CO2的插入固定是反应的决速步,反应按途径B进行在能量上是最有利的.前线分子轨道理论分析进一步验证了计算的结果.
参考文献:
[1]刘霜霜.(PNN)Ru(H)(CO)和(NNS)Ru(H)(CO)活化二氧化碳机理的理论研究[D].临汾:山西师范大学,2014.
[2]潘星.锌系催化剂催化二氧化碳/环氧化合物共聚机理研究[D].上海:华东理工大学,2013.
[3]王勤.若干金属配合物活化二氧化碳机理的理论研究[D].临汾:山西师范大学,2015.
[4]魏俊明.铁化合物的设计、合成、表征及其催化二氧化碳与环氧烷化合物偶联合成环状碳酸醋的研究[D].南昌:南昌大学,2013.
[5]MA R,HE L N,ZHOU Y B.An efficient and recyclable tetraoxo-coordinated zinc catalyst for the cycloaddition of epoxides with carbon dioxide at atmospheric pressure[J].Green Chem.2016,18:226-231.
[6]PENG J,YANG H J,SONG N N,et al.An effective Ni/Zn catalyst system for the chemical fixation of carbon dioxide with epoxides[J].J.CO2.Util.2015,9:16-22.
[7]LANG X D,YU Y C,HE L N.Zn-salen complexes with multiple hydrogen bonding donor and protic ammonium bromide:Bifunctional catalysts for CO2fixation with epoxides at atmospheric pressure[J].J.Mol.Catal.A-Chem.2016,420:208-215.
[8]DIOS M P,IGLESIAS C A,FERNÁNDEZ A,et al.Effect of Zn/Co initial preparation ratio in the activity of double metal cyanide catalysts for propylene oxide and CO2copolymerization[J].Eur Polym J.2017,88:280-291.
[9]ADOLPH M,ZEVACO T A,ALTESLEBEN C,et al.New zinc catalysts based on easy-to-handle N4-chelating ligands for the coupling reaction of epoxides with CO2[J].J.Mol.Catal.A-Chem.2015,400:104-110.
[10]ZHANG X H,WEI R J,ZHANG Y Y,et al.Carbon dioxide/epoxide copolymerization via a nanosized Zinc-Cobalt(iii)double metal cyanide complex:Substituent effects of epoxides on polycarbonate selectivity,regioselectivity and glass transition temperatures[J].Macromolecules.2015,48(3):536-544.
[11]LIU M,LIU B,LIANG L,et al.Design of bifunctional NH3I-Zn/SBA-15 single-component heterogeneous catalyst for chemical fixation of carbon dioxide to cyclic carbonates[J].J.Mol.Catal.A-Chem.2016,418-419:78-85.
[12]WANG F,XU C,LI Z,et al.Mechanism and origins of enantioselectivity for[BMIM]Cl ionic liquids and ZnCl2co-catalyzed coupling reaction of CO2with epoxides[J].J.Mol.Catal.A-Chem.2014,385:133-140.
[13]WANG L,HUANG T,CHEN C,et al.Mechanism of hexaalkylguanidinium salt/zinc bromide binary catalysts for the fixation of CO2with epoxide:ADFT investigation[J].J.CO2.Util.2016,14:61-66.
[14]HUAY Z,YANG X C,LIU M M,et al.Asymmetric copolymerization of cyclopentene oxide and CO2using a dinuclear Zinc-Aze phenol catalyst:enlightened by DFT calculations[J].Macromolecules.2015,48(6):1651-1657.
[15]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09[CP],Revision E.01,Gaussian Inc.,Wallingford CT,2013.
