苯肼作为引发剂的碱促进均裂芳环取代反应研究
2018-03-30,,,
,,,
(安徽工程大学 生物与化学工程学院,安徽 芜湖 241000)
联苯结构存在于众多有生物活性的自然产物中,这些结构可以作为潜在的功能材料或者药物的重要成分来研究[1-5].过渡金属催化的偶联反应是前些年研究的重点,但是这类反应却有着无法逃避的缺点,比如,毒性和大量的金属废弃物[1-3].虽然目前对过渡金属催化偶联反应的研究还是很多,但也出现了更为有价值的碱催化均裂芳环取代反应(Base Promoted Homolytic Aromatic Substitution,BHAS)的方法来研究得到相应的目标产物.两种催化方法的对比如图1所示.这类反应一般有单电子转移(Single Electron Transfer,SET)的过程,经历了自由基反应的历程.这种反应类型的发现为联苯化合物的合成提供了新的方法,也体现了绿色化学发展的趋势[4-10].

图1 两种催化方法的对比图
1 材料及方法
1.1 材料
试剂、药品均为直接购买并且没有进一步除杂提纯.
1.2 方法
利用气相色谱(Agilent 7890A)及气质联用(Trace DSQ)的方法来跟踪反应,分离出来的产物用核磁共振氢谱碳谱结合质谱来定性产物.
2 结果与讨论
2.1 反应机理的分析
由于最近双胺配体结合叔丁醇钾的高效催化组合的出现,BHAS反应受到了极大关注.以下是BHAS反应的机理图示,其中芳基自由基加到苯肼衍生物上形成相应的六元环状自由基,在碱的作用下进一步得到联苯衍生物自由基阴离子;再通过电荷转移和起始原料作用,得到最终产品和卤代芳烃自由基负离子,由此生成的卤代芳烃自由基负离子随后又参与了催化循化,反应机理如图2所示.

图2 反应机理分析
2.2 模板反应
反应以对苯与甲基碘苯的反应来考察不同的反应条件.反应在温度为100 ℃,碱为叔丁醇钾的情况下,考察了催化剂为PhNHNH2的反应用量,发现产率随催化剂的用量的减少而降低,但10 mol%为较经济的用量;也考察了模板反应用不同碱的情况,发现叔丁醇钾是较理想的用碱;变换反应温度,当温度为85 ℃时,反应得到的产物产率明显下降;也考察了不同的催化剂对模板反应的影响,发现苯肼是较为理想的催化剂.据此,确定了反应的最佳反应条件:叔丁醇钾为碱,10 mol%的苯肼为催化剂和100 ℃的反应温度.模板反应的最佳条件选择如表1所示.
表1模板反应最佳条件选择a


编号碱催化剂/mol%温度/℃反应产率b/%1KOt-BuPhNHNH2(40)100852KOt-BuPhNHNH2(20)100833KOt-BuPhNHNH2(10)10084(73)4KOt-BuPhNHNH2(5)100815KOt-BuPhNHNH2(3)100316KOt-BuPhNHNH2(10)85627NaOt-BuPhNHNH2(10)100Trace8LiOt-BuPhNHNH2(10)100Trace9KOHPhNHNH2(10)100Trace10K2CO3PhNHNH2(10)100Trace11KOt-BuPhN(Me)NH2(10)100Trace12KOt-BuPhNHNMe2(10)100Trace13KOt-BuPhNHNHPh(10)10070
注:a对甲基碘苯0.5 mmol,碱2 mmol,苯5 mL,bGC产率(内标法)
2.3 催化体系的底物拓展以及机理讨论
更多的卤代芳烃反应底物被纳入考察(表2,1-15),不同的反应底物在较优反应条件下,反应均可以顺利进行.卤素取代位置不同的邻、间、对碘代芳烃也纳入了考察,反应也都能得到较高的产率,这体现出了反应的通适性.反应对溴苯和氯苯也进行了考察,但催化效果并不理想,反应还能得到三联苯产物,并且反应原料为4-Cl-Ph-I,4-Br-Ph-I和4-I-Ph-I时反应都能得到相应三联苯产物,并且产率差别不大(表2,10-12).
为了更加深入地探究反应机理,自由基捕获试验如图3所示,将两种常用的自由基捕获剂1,1'-二苯基乙烯(diyldibenzene)与2,2,6,6-四甲基哌啶-氮-氧化物(Tempo)加入到模板反应中,加入后反应不能顺利地检测出产物的存在,说明模板反应应该存在着自由反应的历程.反应可以以自由基正常的链反应归类,有引发阶段、增长阶段和终止阶段,并且反应的副产物可以由GC-MS断定,为引发剂苯肼得到的苯自由基和产物的结合.反应可能的机理及副产物推测如图4所示.
表2苯肼作为引发剂促进芳基卤化物与苯直接C-H芳基化的底物拓展a
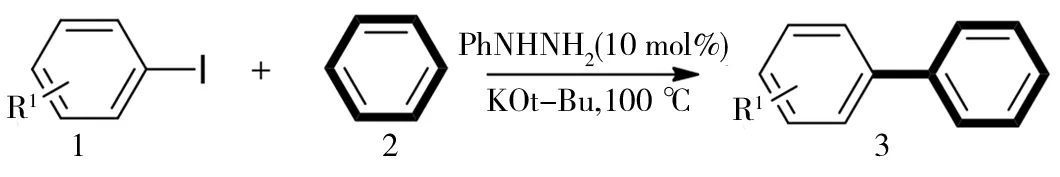
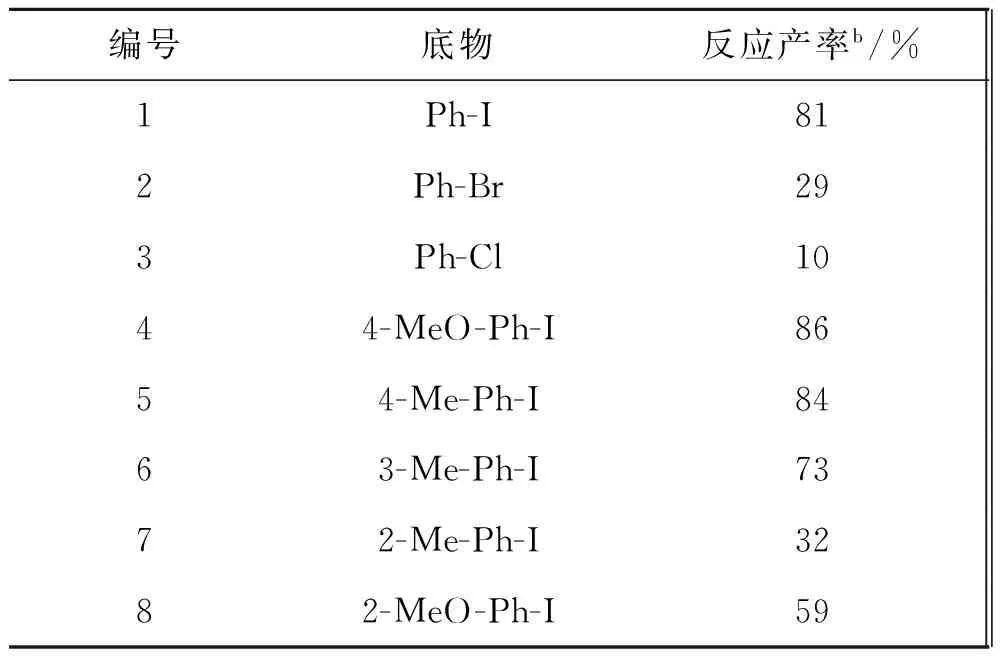
编号底物反应产率b/%1Ph-I812Ph-Br293Ph-Cl1044-MeO-Ph-I8654-Me-Ph-I8463-Me-Ph-I7372-Me-Ph-I3282-MeO-Ph-I59
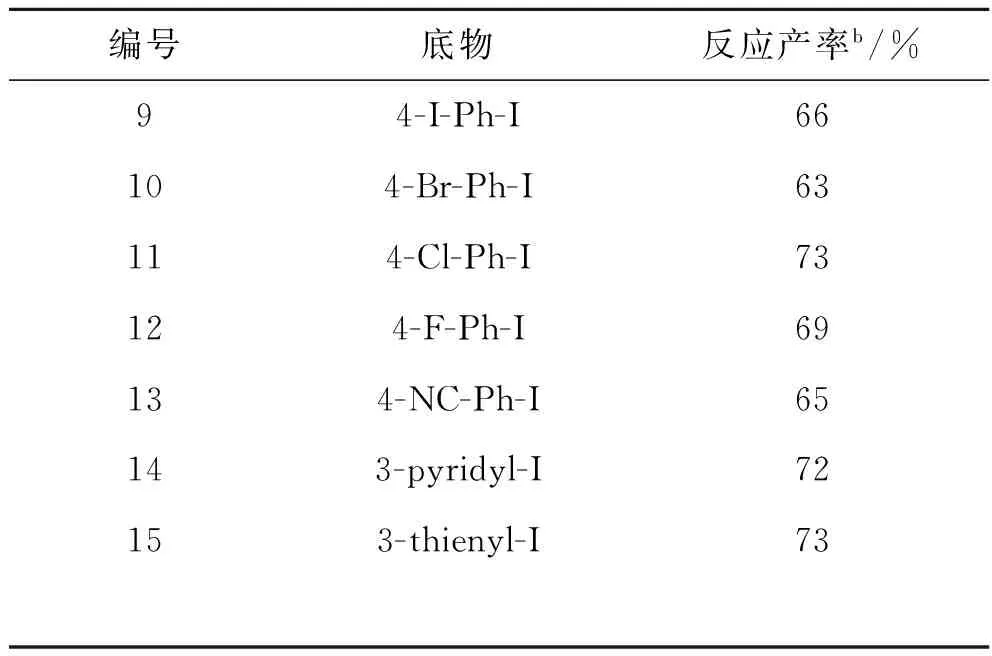
编号底物反应产率b/%94-I-Ph-I66104-Br-Ph-I63114-Cl-Ph-I73124-F-Ph-I69134-NC-Ph-I65143-pyridyl-I72153-thienyl-I73
注:a1(0.5 mmol),PhNHNH2(1.5 mmol),叔丁醇钾(2 mmol),苯(5 mL),100 ℃,bGC产率(内标法)

图3 自由基捕获实验

图4 反应可能的机理及副产物推测
3 结论
以苯肼作为引发剂可以促进无过渡金属参与的偶联反应,可高效地得到相应的偶联反应产物.苯肼经济易得具有现实的合成意义.同时,提出了反应碱促进的均裂芳环取代这一反应机理,并做了相应的机理研究.此研究为以后无过渡金属自由基催化提供了研究思路和实验资料.
[1] A STUDER,D P CURRAN.Organocatalysis and C-H activation meet radical-and electron-transfer reactions[J].Angewandte Chemie International Edition,2011,50(22):5 018-5 022.
[2] T NOEL,T J MAIMONE,S L BUCHWALD.Accelerating Pd-catalyzed C-F bond formation:use of a microflow packed-bed reactor[J].Angewandte Chemie International Edition,2011,50(38):8 900-8 903.
[3] S YANAGISAWA,K UEDA,T TANIGUCHI,et al.Potassium T-butoxide alone can promote the biaryl coupling of electron-deficient nitrogen heterocycles and haloarenes[J].Organic Letters,2008,10(20):4 673-4 676.
[4] Y QIU,Y LIU,K YANG,et al.New ligands that promote cross-coupling reactions between aryl halides and unactivated arenes[J].Organic Letters,2011,13(14):3 556-3 559.
[5] C L SUN,H LI,D G YU,et al.An Efficient organocatalytic method for constructing biaryls through aromatic C-H activation[J].Nature Chemistry,2010,2(12):1 044-1 049.
[6] E SHIRAKAWA,K ITOH,T HIGASHINO,et al.Tert-butoxide-mediated arylation of benzene with aryl halides in the presence of a catalytic 1,10-phenanthroline derivative[J].Journal of the American Chemical Society,2010,132(44):15 537-15 539.
[7] N UCHIYAMA,E SHIRAKAWA,R NISHIKAWA,et al.Iron-catalyzed oxidative coupling of arylboronic acids with benzene derivatives through homolytic aromatic substitution[J].Chemical Communications,2011,47(42):11 671-11 673.
[8] J WEN,J ZHANG,S Y CHEN,et al.Iron-mediated direct arylation of unactivated arenes[J].Angewandte Chemie International Edition,2008,47(46):8 897-8 900.
[9] D LIU,C LIU,H LI,et al.Direct functionalization of tetrahydrofuran and 1,4-dioxane:nickel-catalyzed oxidative C(sp3)-H arylation[J].Angewandte Chemie International Edition.,2013,52(16):4 453-4 456.
[10] J A MURPHY.Discovery and development of organic super-electron-donors[J].The Journal of Organic Chemistry,2014,79(15):3 731-3 746.
