Prader-Willi综合征两例报道并文献复习
2018-03-21魏雯涂梅陈彤陈阳
魏雯,涂梅,陈彤,陈阳
Prader-Willi综合征(PWS)又称隐睾-侏儒-肥胖-智力低下综合征和肌张力低下-智能障碍-性腺发育滞后-肥胖综合征,最早由PRADER等[1]于1956年报道而得名,是导致人类肥胖的最常见遗传性综合征之一。PWS是一种累及多系统的非孟德尔遗传性疾病,是印迹遗传的典型代表[2],国外报道发病率为1/(10 000~30 000)[3-4]。早发现、早治疗有助于改善疾病预后,但该疾病早期临床表现不典型,早期诊断有一定困难。本文结合2例青春期PWS患儿进行报道,以期提高临床对该病的认识。
1 病例简介
患儿1,男,13岁,因体质量进行性增长9年,于2014-08-08入住福建医科大学附属龙岩第一医院。患儿足月顺产,出生时身长50 cm,体质量3 kg,出生时因舌系带过短行手术治疗。出生后人工汤匙喂养,进食量少,少哭闹,4岁时学会走路、说话,上幼儿园至今不爱运动。4岁前有反复发热病史。4岁后食欲好,食量大,体质量进行性上升,约8 kg/年,6年前(7岁)曾就诊福州儿童医院,完善相关检查,自诉未见明显异常(未见检查单),平素喜抓挠皮肤,智力较同龄人低下,就读于特殊学校。父母非近亲结婚,无家族性遗传病史。父亲43岁,身高156 cm,母亲45岁,身高157 cm。入院查体:体温37.3 ℃,脉搏106次/min,呼吸18次/min,血压100/70 mm Hg(1 mm Hg=0.133 kPa),身高138 cm,体质量78 kg,体质指数(BMI)41.0 kg/m2,体型肥胖,反应迟钝,颈项部黑棘皮表现,皮肤见抓痕。面色白,杏仁眼,甲状腺无肿大,男性乳腺发育,腹部皮肤多处紫纹,手足较小。无腋毛、阴毛,阴茎长约2 cm,阴茎Tanner分期1期,睾丸未触及。辅助检查:三酰甘油3.44 mmol/L(参考范围:0.39~1.10 mmol/L),低密度脂蛋白胆固醇3.27 mmol/L(参考范围:1.30~3.15 mmol/L),尿酸492 μmol/L(参考范围:90~420 μmol/L),糖化血红蛋白6.3%(参考范围:3.6%~6.0%),空腹血糖5.06 mmol/L,胰岛素318.7 pmol/L(参考范围:17.8~173.0 pmol/L),C肽2.16 nmol/L(参考范围:0.37~1.47 nmol/L),餐后2 h血糖6.39 mmol/L,餐后2 h胰岛素1 515 pmol/L,餐后2 h C肽5.90 nmol/L。甲状腺功能:游离三碘甲腺原氨酸(FT3)6.76 pmol/L,游离甲状腺素(FT4)15.80 pmol/L,促甲状腺激素(TSH)2.59 mU/L。皮质醇节律:410.2 nmol/L(8:00)、249.2 nmol/L(16:00)、62.60 nmol/L(次日0:00)。黄体生成素(LH)0.428 mU/ml(参考范围:1.700~8.600 mU/ml),卵泡刺激素(FSH)0.217 mU/ml(参考范围:1.500~12.400 mU/ml),睾酮<0.087 nmol/L(参考范围:9.900~27.800 nmol/L),人绒毛膜促性腺激素(HCG)兴奋试验:睾酮0.217 nmol/L,生长激素1.19 μg/L,精氨酸兴奋试验:30 min 1.75 μg/L、60 min 3.47 μg/L、90 min 2.45 μg/L、120 min 2.91 μg/L。阴囊彩超:右侧睾丸未探及,左侧睾丸体积偏小。乳腺彩超:双侧乳腺发育。腹部彩超:脂肪肝。心脏彩超:右室增大。鞍区CT:鼻咽顶部软组织增厚,腺样体肥大可能,鞍区未见异常。染色体核型分析:46XY。经上海交通大学医学院附属新华医院基因检测,患儿DNA模板经McrBC消化后,SNRPN序列不能扩增;经HpaⅡ消化后,SNRPN序列能扩增,结果与PWS阳性参照一致(见图1)。给予生活方式、运动等干预,并予二甲双胍500 mg,3次/d,随访6个月,患儿体质量下降10 kg。
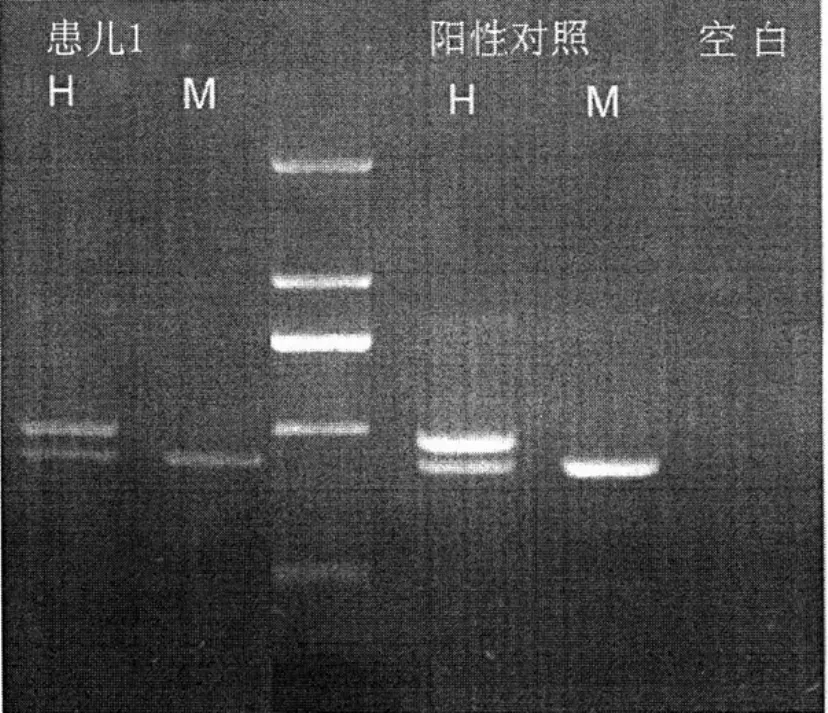
图1 患儿1基因检测结果Figure 1 Genetic testing results of No.1 patient(a 13-year-old male) with Prader-Willi syndrome
患儿2,女,14岁,因体质量进行性增长10年,于2015-08-18入住福建医科大学附属龙岩第一医院。患儿于母亲妊娠期间胎动较弱,足月难产剖宫产,出生时有缺氧,出生时身长60 cm,体质量2.5 kg,曾患“新生儿肺炎”。出生后人工喂养,120 ml奶需食用1 h,少哭闹,6个月抬头,8个月出牙,12个月说话,18个月走路,4岁言语尚流利,上幼儿园至今不爱运动,一直尿床至12岁左右。4岁时因臀部脓肿,治疗1年。4岁后食欲好,食量大,喜食零食、易饥饿。体质量以5~10 kg/年的速度增长,身高增长2~3 cm/年,近1年身高增长停止。学习成绩一般,性格偏执,自律性差,平素喜抓挠皮肤。12岁时阴毛、腋毛开始出现,无月经来潮。1年前就诊济宁医学院附属医院,诊断“原发性甲状腺功能减退”,T4正常,TSH 4.97 mU/L,予“左旋甲状腺素3/4片”治疗。2015年2、3月曾予“二甲双胍500 mg,3次/d”治疗,体质量无下降,月经仍未来潮。父母体健,非近亲结婚,父亲43岁,身高168 cm,母亲41岁,身高158 cm。入院查体:体温36.5 ℃,脉搏78次/min,呼吸20次/min,血压120/80 mm Hg,身高150 cm,体质量98 kg,BMI 43.6 kg/m2。反应迟钝,言语欠清晰,胸背部色素沉着,皮肤见抓痕。面色白,眼裂偏小,甲状腺无肿大。腹部见白纹,四肢毳毛增多增粗,手足较小。乳房Tanner分期1期,无腋毛,会阴毛发呈女性分布,阴毛Tanner分期3期。辅助检查:糖化血红蛋白5.6%,空腹血糖4.68 mmol/L,胰岛素17.63 μU/ml(参考范围:1.25~10.50 μU/ml),C肽4.82 μg/L(参考范围:0.82~2.50 μg/L)。餐后2 h血糖6.81 mmol/L,餐后2 h胰岛素122.40 μU/ml,餐后2 h C肽15.78 μg/L。甲状腺功能:FT34.58 pmol/L(参考范围:3.10~6.80 pmol/L),FT411.32 pmol/L(参考范围:12.00~22.00 pmol/L),TSH 5.76 mU/L(参考范围:0.27~4.20 mU/L),抗甲状腺球蛋白抗体16.66 kU/L,抗甲状腺过氧化物酶抗体16.09 kU/L。皮质醇节律 7.12 μg/dl(8:00)、5.21 μg/dl(16:00)、3.48 μg/dl(次日0:00),1 mg地塞米松抑制后次日皮质醇0.59 μg/dl(8:00),24 h 尿皮质醇:106.78 μg。LH<0.070 U/L(参考范围:卵泡期1.900~12.500 U/L),FSH 0.510 U/L(参考范围:卵泡期2.500~10.200 U/L),雌二醇139.55 pmol/L(参考范围:卵泡期71.60~529.20 pmol/L),戈那瑞林试验:LH最高值0.37 U/L。生长激素0.06 μg/L,胰岛素低血糖试验:血糖1.23 mmol/L时生长激素为5.988 μg/L。左手骨龄片:骨龄略大于实际年龄。盆腔MR:子宫形态小,宫颈内可疑纵行分隔;左侧附件区结节状信号影,不除外卵巢结构可能;右侧附件未见确切显示。染色体核型分析:46XX。肾上腺CT平扫、鞍区MR:未见明显异常。甲状腺彩超:甲状腺未见异常。腹部彩超:脂肪肝。喉镜:鼻炎、扁桃体肥大、咽喉炎。该患者家属拒绝行基因检测,根据HOLM等[5]于1993年提出和CASSIDY等[3]于2012年修正后的标准,临床诊断PWS成立。治疗上予生活方式、运动等干预,并予二甲双胍500 mg,3次/d,左旋甲状腺素片50 μg,1次/d,随访3个月,患儿体质量下降8 kg。
2 讨论
PWS是一种累及多系统的疾病,目前临床诊断主要参照HOLM等[5]于1993年提出,CASSIDY等[3]于2012年修正后的标准,包括6条主要标准、11条次要标准和8条支持证据。主要标准每项1分,次要标准每项0.5分。>3岁总分超过8分(主要标准得分≥5分)即可诊断。患者1评分9分,主要标准得分6分,患者2评分10分,主要标准得分6分,临床诊断成立。该综合征随年龄变化其临床表现有所不同。新生儿、婴儿期以肌张力低下为突出表现;儿童期以生长缓慢和肥胖为主,面部可有特殊面容,表现为小双额径、杏仁眼、三角嘴;青春期则有肥胖、心理障碍、青春期发育滞后、性腺发育不全,易诱发青春期糖尿病。2例患儿均经历了典型的PWS发展过程:出生后少哭闹,吸吮力差,生长发育均落后于同龄儿。4岁出现摄食过度至体质量迅速增长,生殖器官发育不全,另有杏仁眼、小手小脚等特征性外观。入院后检查提示生长激素缺乏,LH、FSH、性激素均明显低下,胰岛素抵抗、脂肪肝、扁桃体、腺样体肥大等。但2例患儿在婴儿期、儿童期均未得到充分认识,在青春期明显肥胖时才得到确诊,因而错过了最佳治疗时间。
PWS的发病机制是染色体15q11.2-13基因组印迹遗传异常,可由4种不同的缺陷类型所致[6]:(1)父源染色体15q11.2-13片段缺失(西方65%~75%,中国和亚洲人群80%[7]);(2)母源同源二倍体(20%~30%);(3)印记中心微缺失及突变(1%~3%);(4)染色体平衡易位(<1%)。因此,确诊需基因学检查,甲基化特异性聚合酶链反应(MS-PCR)为目前首选方法,检出率≥99%,但无法区分各种缺陷类型[8]。因绝大多数染色体微缺失和母源性单亲二倍体是偶然发生的,患者父母再次生育的复发风险很低,约为1%,若为印记中心突变,则复发风险高,为50%[3],所以缺陷类型的检查对再发风险的估计至关重要。若患者父母有再次生育的要求,就需进一步分析缺陷类型。此外,还可采用MS-PCR通过羊水脱落细胞在妊娠16~20周进行产前诊断[9]。本研究患者1基因学检查结果阳性,可明确诊断,父母暂时无再次生育要求,故未进一步行缺陷类型分析。
PWS的治疗应根据不同年龄段患者的特征,针对内分泌代谢紊乱和心理行为问题进行综合干预。本研究报道的2例患儿均为青春期年长儿,存在难以自控的摄食行为,是造成肥胖的原因,进而引发糖尿病、血脂异常、梗阻性呼吸困难、心力衰竭等。对于PWS患者,如果能避免肥胖,其寿命可以接近正常,国外文献报道PWS死亡患者年龄最大为68岁[10],因此控制饮食、减轻体质量和体育锻炼等行为治疗是十分重要的。但此类患者均有一定程度的智力障碍和无法自控的摄食行为,患者亲属需严格管理食物,制定三餐计划[9]。本研究2例患儿通过控制饮食、体育锻炼等行为治疗,并予二甲双胍坚持治疗,体质量均有不同程度的下降。二甲双胍作为一种半百老药,可减少脂肪合成、改善胰岛素抵抗、减少食欲、增加饱腹感,达到减重效果,因此,可作为饮食、运动和行为治疗的辅助。MILLER等[11]对21例PWS患儿给予二甲双胍治疗,其中5例开始能感受到饱足感,并且食物相关的紧张、焦虑情绪和控制自身远离食物的能力均有所改善,其机制尚不十分清楚,可能与糖耐量和高胰岛素血症改善相关。因此,笔者认为,二甲双胍性价比高,若无禁忌证,建议二甲双胍配合行为治疗长期使用。
目前还有报道代谢手术和胰高血糖素样肽1(GLP-1)治疗PWS合并糖尿病患者的病例[12-13],也获得一定的疗效。但本研究2例患儿无糖尿病,且未满18岁,暂不适用。而代谢手术仅缩小了胃容积,得到了一定的近期疗效,患儿无法自控的摄食行为仍存在,故远期疗效不明确。新近有报道减肥新药贝洛雷尼开始Ⅲ期临床试验,此药可降低肝脂肪合成和身体的脂肪贮存,可减轻饥饿感,促进脂肪代谢[14]。对于生长激素治疗方面,一般认为初治时间为婴幼儿早期、肥胖发生前(2岁前)[15],早期治疗可改善患儿精神运动发育。治疗可一直持续至成年期,即使骨骺融合仍有改善脂代谢和认知功能的作用[9]。但严重肥胖、未控制的糖尿病、未控制的严重睡眠呼吸暂停综合征、活动性肿瘤和活动性精神病禁用重组人生长激素[16]。本研究2例患儿均严重肥胖,且合并扁桃体、腺样体肥大等问题,因此不适于重组人生长激素治疗。对于其他内分泌问题,如病例2存在甲状腺功能减退,应予左旋甲状腺素片替代治疗,根据T4和FSH水平调整剂量[9]。本研究2例患儿均错过了最佳重组人生长激素治疗时间,因此,临床医生尤其是儿科医生应加强对本病的认识,尽可能在早期给予诊断,让患儿在肥胖出现前尽早得到治疗,改善患儿预后。
总之,Prader-Willi综合征临床表现多样,随着年龄进展而呈现不同的特点,预后不良,迄今为止尚无治愈方法,以对症治疗为主。对于青春期患儿,应加强饮食和运动管理,若无禁忌证,二甲双胍应是首选,可联合胰高血糖素样肽1(GLP-1)类似物治疗。生长激素的治疗应早期进行,且应充分权衡利弊,还需对甲状腺功能异常等其他内分泌激素改变进行相应的处理。随着医学研究的不断深入,期待贝洛雷尼等减肥新药能尽早使用于临床,造福于患者。而最关键的是,临床医生应加强对该病的认识,对患儿肥胖出现之前比如肌张力低下、生长发育迟缓、特殊面容等特征能有充分的认识,才能做到早期诊断和治疗,切实造福于患者。
作者贡献:魏雯进行文章的构思与设计、文献/资料收集整理、撰写论文;陈阳进行文献/资料整理;陈彤负责文章的质量控制及审校;涂梅对文章整体负责,监督管理。
本文无利益冲突。
[1]PRADER A,LABHART A,WILLI H.Ein syndrome von Adipositas,Kleinwuchs,Kryptorchismus und Oligophreniee nach myatonieartigerm Zustand im Neugeborenenalter[J].Schweiz Med Wochenschr,1956,86:1260-1261.
[2]MCCANDLESS S E,Committee on Genetics.Clinical reporthealth supervision for children with Prader-Willi syndrome[J].Pediatrics,2011,127(1):195-204.DOI:10.1542/peds.2010-2820.
[3]CASSIDY S B,SCHWARTZ S,MILLER J L,et al.Prader-Willi syndrome[J].Genet Med,2012,14(1):10-26.DOI:10.1038/gim.0b013e31822bead0.
[4]BUTLER M G.Prader-Willi syndrome:obesity due to genomic imprinting[J].Curr Genomics,2011,12(3):204-215.DOI:10.2174/138920211795677877.
[5]HOLM V A,CASSIDY S B,BUTLER M G,et al.Prader-Willi syndrome:consensus diagnostic criteria[J].Pediatrics,1993,91(2):398-402.
[6]CRINO A,SCHIAFFINI R,CIAMPALINI P,et al.Hypogonadism and pubertal development in Prader-Willi syndrome[J].Eur J Pediatr,2003,162(5):327-333.DOI:10.1007/s00431-002-1132-4.
[7]LU W,QI Y,CUI B,et al.Clinical and genetic features of Prader-Willi syndrome in China[J].Eur J Pediatr,2014,173(1):81-86.DOI:10.1007/s00431-013-2124-2.
[8]朱丽娜,何玺玉,王春枝,等.Prader-Willi综合征的分子遗传学诊断与机制研究[J].山西医科大学学报,2008,39(12):1064-1067.DOI:10.3969/j.issn.1007-6611.2008.12.003.ZHU L N,HE X Y,WANG C Z,et al.Genetic diagnosis and mechanism of Prader-Willi syndrome[J].Journal of Shanxi Medical University,2008,39(12):1064-1067.DOI:10.3969/j.issn.1007-6611.2008.12.003.
[9]中华医学会儿科学分会内分泌遗传代谢学组,《中华儿科杂志》编辑委员会.中国Prader-Willi综合征诊治专家共识(2015)[J].中华儿科杂志,2015,53(6):419-424.DOI:10.3760/cma.j.issn.0578-1310.2015.06.005.The Subspecialty Groups of Endocrine Genetic Metabolism,the Society of Pediatrics,Chinese Medical Association,Editorial Board of Chinese Journal of Pediatrics.Consensus of experts on the diagnosis and treatment of Prader-Willi syndrome in China(2015)[J].Chinese Journal of Pediatrics,2015,53(6):419-424. DOI:10.3760/cma.j.issn.0578-1310.2015.06.005.
[10]SCHRANDER-STUMPEL C T,CURFS L M,SASTROWIJOTO P,et al.Prader-Willi syndrome:causes of death in an international series of 27 cases[J].Am J Med GenetA,2004,124A(4):333-338.DOI:10.1002/ajmg.a.20371.
[11]MILLER J L,LINVILE T D,DYKENS E M.Effects of metformin in children and adolescents with Prader-Willi syndrome and early-onset morbid obesity:a pilot study[J].J Pediatr Endocrinol Metab,2014,27(1/2):23-29.DOI:10.1515/jpem-2013-0116.
[12]吴佳君,乔洁,韩兵,等.一例Prader-Willi综合征的基因诊断和减重手术治疗[J].中华内分泌代谢杂志,2011,27(6):498-501.DOI:10.3760/cma.j.issn.1000-6699.2011.06.014.WU J J,QIAO J,HAN B,et al.Genetic diagnosis and weight loss surgery of a case of Prader-Willi syndrome[J]. Chinese Journal of Endocrinology and Metabolism,2011,27(6):498-501.DOI:10.3760/cma.j.issn.1000-6699.2011.06.014.
[13]许建萍,肖新华.Prader-Willi综合征的代谢异常及胰高血糖素样肽1类似物治疗[J].中华糖尿病杂志,2014,6(6):427-429.DOI:10.3760/cma.j.issn.1674-5809.2014.06.017.XU J P,XIAO X H.Metabolic abnormalities and treatment of glucagon like peptide 1 analog of Prader-Willi syndrome[J].Chinese Journal of Diabetes Mellitus,2014,6(6):427-429.DOI:10.3760/cma.j.issn.1674-5809.2014.06.017.
[14]减肥新药贝洛雷尼治疗Prader-Willi综合征[J].国际药学研究杂志,2014,41(6):666.Beloranib,a new weight-loss drug for the treatment of Prader-Willi syndrome[J].Journal of International Pharmaceutical Research,2014,41(6):666.
[15]中华医学会儿科学分会内分泌遗传代谢学组,《中华儿科杂志》编辑委员会.基因重组人生长激素儿科临床规范应用的建议[J].中华儿科杂志,2013,51(6):426-432.DOI:10.3760/cma.j.issn.0578-1310.2013.06.007.The Subspecialty Groups of Endocrine Genetic Metabolism,the Society of Pediatrics,Chinese Medical Association,Editorial Board of Chinese Journal of Pediatrics. Proposal for the application of recombinant human growth hormone in pediatric clinical practice[J].Chinese Journal of Pediatrics,2013,51(6):426-432. DOI:10.3760/cma.j.issn.0578-1310.2013.06.007.
[16]DEAL C L,TONY M,HOYBYE C,et al.Growth Hormone in Prader-willi Syndrome Clinical Care Guidelines Workshop Participants.Growth Hormone Research Society workshop summary:consensus guidelines for recombinant human growth hormone therapy in Prader-Willi syndrome[J].J Clin Endocrinol Metab,2013,98(6):E1072-1087.DOI:10.1210/jc.2012-3888.
