基于反转录转座子及简单重复序列标记的裸大麦遗传多样性研究
2018-03-21王国荣陈功海龙周锴张文英徐延浩
王国荣,华 为,陈功海,龙周锴,李 博,张文英,徐延浩,*
(1.主要粮食作物产业化湖北省协同创新中心/长江大学 农学院,湖北 荆州 434025; 2.浙江省农业科学院 作物与核技术利用研究所,国家大麦改良中心,浙江 杭州 310021; 3.荆州市农业科学院,湖北 荆州 434000)
裸大麦(青稞)是青藏高原地区最重要的粮食作物,但其育成品种与种质资源材料的遗传多样性水平存在争议。孟凡磊等[1]、杨平等[2]的研究认为,川藏地区育成青稞品种的遗传多样性较低。曾兴权等[3]的研究表明,同一来源地区的青稞育成品种遗传基础较为狭窄,但不同地区间的遗传差异较大。Feng等[4]、赖勇等[5]和巴桑玉珍等[6]利用简单重复序列(simple sequence repeat,SSR)标记对青藏地区青稞品种与资源材料的群体遗传结构、地理分化及与重要农艺性状的关联分析进行研究,认为青稞育成品种遗传基础较为狭窄,但种质资源的遗传多样性较高。
反转录转座子是高等植物基因组中广泛存在的一类可移动的DNA元件[7],是植物基因组进化的重要动力[8-10],参与植物性染色体形成[10]。反转录转座子是大麦基因组的重要组成部分[11],是大麦基因组响应外界胁迫的重要元件[12-13],并能介导基因缺失[13],在大麦基因组进化中具有重要作用[13-14]。反转录转座子标记具有灵敏度高、基因组覆盖度广、多态性高等特点,适用于遗传多样性研究及种质资源鉴定[15-17]。利用分子标记进行遗传多样性分析、指纹图谱构建、关联分析研究的报道在动植物中都较为常见[18-20],然而利用反向反转录转座子扩增多态性(inter-retrotransposon amplified polymorphism,IRAP)、反转录转座子-微卫星扩增多态性(retrotransposon-microsatellite amplified polymorphism,REMAP)等反转录转座子标记进行大麦遗传群体结构研究的报道较少[17],至今未见利用IRAP、REMAP揭示裸大麦育成品系及资源材料遗传多样性和遗传结构的报道。
鉴于此,本研究选取63份来自不同地区的裸大麦品系及西藏裸大麦资源材料,利用10对REMAP标记、13对IRAP标记对这些材料的遗传多样性及群体遗传结构进行研究,并与SSR标记揭示的遗传多样及群体结构进行比较分析,从反转录转座子角度丰富裸大麦的遗传多样性研究,提供新颖的遗传多样性资料,以期更好地解析裸大麦的遗传基础,为裸大麦及其种质资源的高效利用提供更多依据。
1 材料与方法
1.1 试验材料
供试63份裸大麦材料包含36份青海品系、9份西藏野生资源材料、4份西藏品系、5份云南品系、3份江苏品系、2份国外材料,以及湖北、甘肃、黑龙江、浙江材料各1份(表1)。供试青海、西藏品系由拉萨市农业科学研究所提供。供试西藏野生材料由武汉大学生命科学学院丁毅教授提供。其他材料由浙江省农业科学院作物与核技术利用研究所国家大麦改良中心提供。
PCR体系中所用10×buffer、Mg2+、dNTPs、TaqDNA聚合酶均购自上海博彩生物科技有限公司,引物由南京金斯瑞生物科技有限公司合成。
1.2 DNA提取
取三叶期新鲜叶片,用CTAB小样法提取基因组DNA,琼脂糖凝胶电泳检测所提DNA的质量,NanoDrop 2000c超微量分光光度计测定DNA浓度,-80 ℃保存备用。
表1供试裸大麦材料的名称和来源
Table1Name and origin of hulless barley accessions used in this study
编号名称来源编号名称来源No.NameOriginNo.NameOrigin1东北186Dongbei186黑龙江Heilongjiang33北青2Beiqing2青海Qinghai2甘青4Ganqing4甘肃Gansu34北青3Beiqing3青海Qinghai308⁃23476国外品系Foreignspecies35北青5Beiqing5青海Qinghai4鄂507Edamai507湖北Hubei36北青6Beiqing6青海Qinghai5苏裸1号Suluo1江苏Jiangsu37门农1Mennong1青海Qinghai6苏裸2号Suluo2江苏Jiangsu38互青Huqing青海Qinghai7南京3Nanjing3江苏Jiangsu39福8⁃4Fu8⁃4青海Qinghai8Inbyf⁃08⁃11欧洲Europe40福8⁃6Fu8⁃6青海Qinghai9紫点裸Zidianluo青海Qinghai41高原早1号Gaoyuanzao1青海Qinghai10紫黑青裸Ziheiqingluo青海Qinghai42昆仑1Kunlun1青海Qinghai11青海1Qinghai1青海Qinghai43昆仑3Kunlun3青海Qinghai12青海2Qinghai2青海Qinghai44昆仑10Kunlun10青海Qinghai13青海3Qinghai3青海Qinghai45短芒青裸Duanmangqingluo西藏Tibet14青海4Qinghai4青海Qinghai46藏青26Tibet26西藏Tibet15青海5Qinghai5青海Qinghai47仁比16Renbi16西藏Tibet16青海6Qinghai6青海Qinghai481574∗西藏Tibet17青海7Qinghai7青海Qinghai491578∗西藏Tibet18青海8Qinghai8青海Qinghai501579∗西藏Tibet19青海9Qinghai9青海Qinghai511585∗西藏Tibet20青海10Qinghai10青海Qinghai521598∗西藏Tibet21青海11Qinghai11青海Qinghai5380284∗西藏Tibet22青海12Qinghai12青海Qinghai5480315∗西藏Tibet23青海13Qinghai13青海Qinghai5580322∗西藏Tibet24青海15Qinghai15青海Qinghai5680402∗西藏Tibet25青海16Qinghai16青海Qinghai57藏320Tibet320西藏Tibet26青海17Qinghai17青海Qinghai58紫光芒166Ziguangmang166云南Yunnan27青海18Qinghai18青海Qinghai59黑麦10号Rye10云南Yunnan28青海19Qinghai19青海Qinghai60紫光芒Ziguangmang云南Yunnan29青海21Qinghai21青海Qinghai61云香Yunxiang云南Yunnan30青海130Qinghai130青海Qinghai62云裸1号Yunluo1云南Yunnan31青海132Qinghai132青海Qinghai63萧山立夏黄Xiaoshanlixiahuang浙江Zhejiang32北青1Beiqing1青海Qinghai
标*的系野生资源。
* indicated wild accessions.
1.3 反转录转座子标记分析
IRAP与REMAP标记引物序列见表2。用于IRAP标记的共有13对组合,分别为A1、A2、A3、A4、A10、A1+A11、A2+A11、A6+A13、A12+A13、A1+A2、A1+A5、A12+A14、A13+A14;用于REMAP标记的共有10对组合,分别为A2+A15、A5+A16、A5+A15、A6+A16、A9+A15、A11+A16、A12+A15、A13+A15、A14+A15、A2+A16。
PCR扩增体系(20 μL):10×buffer 2 μL,25 mmol·L-1Mg2+1.6 μL,10 mmol·L-1dNTPs 0.4 μL,5 U·μL-1Taq酶0.3 μL,10 mmol·L-1引物各1 μL,50 ng·μL-1模板DNA 2 μL,ddH2O 11.7 μL。PCR扩增程序:94 ℃预变性5 min;94 ℃变性30 s,54~58 ℃退火40 s,72 ℃延伸2 min,36个循环;72 ℃延伸10 min。
REMAP标记PCR产物使用质量分数为2.5%的琼脂糖凝胶电泳检测,使用凝胶成像系统(Syngene, G:BOX-CHEMI-XRQ)记录结果;IRAP标记PCR产物使用质量分数为5%的非变性聚丙烯酰胺凝胶电泳分离,硝酸银染色检测带纹,数码相机拍照记录结果。
表2IRAP和REMAP引物序列
Table2Primer sequences of IRAP and REMAP

编号No.引物名称Name引物序列Primersequence(5′→3′)A1Sukkula⁃E0228GGAACGTCGGCATCGGGCTGA2Sukkula⁃E9900GATAGGGTCGCATCTTGGGCGTGACA3BAGY⁃1⁃C2043TCGTCGCCGGCGTCATCTCCA4BAGY⁃1⁃4164CGATGTGTTACAGGCTGGATTCCA5Nikita⁃57CGGATTTGTTCAAGCCTAAACCA6BARE⁃1⁃5980⁃ACTAGGGCATAATTCCAACAAA7BARE⁃1⁃92460CTGGCTAGCCAACTAGAGGCTTGCA8BARE⁃1⁃E1814TTCCCATGCGACGTTCCCCAACA9BARE⁃1⁃C0700ACACACAAAGCATTCCTCCGGA10Sabrina⁃C0945GCAAGCTTCCGTTTCCGCA11Sukkula⁃91673TGTGACAGCCCGATGCCGACGTTCCA12BARE⁃1⁃5980⁃GCTAGGGCATAATTCCAACGA13BARE⁃1⁃5980⁃ACCCTAGGGCATAATTCCAACACCA14BARE⁃1⁃5980⁃GTCTAGGGCATAATTCCAACGTA15GA9ACGAGAGAGAGAGAGAGAGAACA16GA9CGAGAGAGAGAGAGAGAGAC
1.4 SSR标记分析
SSR标记引物及其染色体位置信息由澳大利亚莫道克大学李承道教授提供(表3)。SSR扩增体系(10 μL):10×buffer 1.0 μL,25 mmol·L-1Mg2+0.8 μL,10 mmol·L-1dNTPs 0.2 μL,5 U·μL-1TaqDNA聚合酶0.15 μL,10 μmol·L-1正反向引物各0.5 μL,50 ng·μL-1模板DNA 2 μL,ddH2O 4.85 μL。SSR标记PCR扩增程序:95 ℃预变性4 min;94 ℃变性30 s,54~61 ℃退火50 s,72 ℃延伸50 s,33个循环;72 ℃延伸10 min。SSR标记PCR产物使用质量分数为6%的非变性聚丙烯酰胺凝胶电泳分离,硝酸银染色检测带纹,数码相机拍照记录结果。
1.5 数据统计分析
IRAP、REMAP和SSR标记的结果以二进制和基因型记录,同一位点上具有相同迁移率的条带记为1,无带记为0,同时以数字1、2、3 等代表不同等位基因。通过Popgene 32统计分析计算基因频率、Shannon指数等遗传参数。以NTSYS-PC软件计算遗传相似性系数(genetic similarity coefficient,GS),按照非加权平均法(unweighted pair-group method with arithmetic means,UPGMA)和SHAN 程序进行聚类分析作图。利用EIGEN程序进行主坐标分析。以Structure 2.3.1软件分析群体遗传结构,估计最佳群体组群数(K),取值范围为1~11,将参数iterations设为10 000次,再将burn-in period设为100 000次,重复11次,计算Q参数。当K值持续增大时,通过计算ΔK确定K值[21]。
2 结果与分析
2.1 标记多态性分析
表3SSR标记染色体位置及引物序列信息
Table3Information of SSR primers
REMAP标记片段大小集中于500~3 000 bp之间(图1-A),10对标记共检测到143个等位变异,每标记平均14.30个,变幅7~25个,其中有131个多态性位点,占总条带数的91.61%,平均多样性指数为0.303 3,每个位点的有效等位基因数为1.514 6。IRAP标记条带大小集中于500~2 000 bp(图1-B),13对标记共检测到315个等位变异,每标记平均24.23个,变幅9~58个,其中有303个多态性位点,占总条带数的96.19%,平均多样性指数为0.268 2,每个位点的有效等位基因数为1.440 5。16对SSR标记共检测到38个等位变异(图1-C),每标记平均2.38个,条带集中于100~400 bp,其中有16个多态性位点,多态性位点百分率 100%,平均多样性指数为0.342 8,每个位点的有效等位基因数为1.610 2。
标记类型不同,单标记所得条带数及有效变异数差异明显。SSR标记单标记平均贡献2.38条条带,平均有效变异数为2.38;REMAP标记单标记平均贡献14.30条条带,平均有效变异数为13.10;IRAP标记单标记平均贡献24.23条条带,平均有效变异数为23.31。较之SSR标记,反转录转座子标记的单一标记多态性位点贡献率较高。
2.2 遗传相似系数及主坐标聚类分析
反转录转座子标记聚类结果显示,63份裸大麦材料GS变幅为0.452~0.937,平均值0.674(图2-A)。其中:紫光芒与紫光芒166间的GS值最大(0.931),表明二者亲缘关系最近;青海21与野生材料80322、80402的GS值最小(均为0.452),表明其亲缘关系较远(图2-A)。在GS值0.620水平上,63份祼大麦材料分为2大类。第1大类可进一步分为2个亚类,第1亚类由鄂507、苏裸2号及Inbyf-08-11组成,第2亚类中包含8份栽培材料和9份野生材料。在GS值0.740水平上,第2亚类中的9份野生材料单独聚为一个小类(图2-A),说明反转录转座子标记可以很好地区分野生裸大麦材料与其他材料。第2大类在GS值0.680水平上以青海1为一个亚类,剩余42份材料为另一亚类。
反转录转座子标记主坐标分析结果显示(图3-A),反转录转座子标记将供试材料分为2大类。第1大类共20份材料包含3个亚类:第1个亚类包含鄂507、苏裸2号、Inbyf-08-11;第2个亚类是9份野生材料;第3个亚类为其余8份材料。第2大类共43份材料,由青海1和其余42份材料组成。主坐标分析结果与聚类分析结果一致度较高,材料所属类群界限明显。
SSR标记聚类结果(图2-B)显示,63份裸大麦材料GS变幅为0.351~0.973,平均值0.716。其中:门农1与青海7的GS值最大(1.000),表明二者亲缘关系较近,其次为北青3与青海7、北青3与门农1,GS值均达到0.973;青海6与苏裸2号的GS值最小(0.351),表明二者亲缘关系较远。聚类分析表明,在GS值0.620水平上63份裸大麦材料可分为2大类,其中苏裸1号、苏裸2号为第1类,其余的61份材料为第2类。第2类在GS值0.660水平上可分为2个亚类,大部分野生裸大麦(8份)包括在第1亚类的21份材料中,裸大麦1579和青海4等40份材料为第2亚类(图2-B)。SSR标记聚类结果中,野生裸大麦材料没有被单独聚为一类。
SSR标记主坐标分析结果(图3-B)显示,SSR标记将供试材料分为2大类:第1大类包括黑麦10号等22份材料;第2大类包含2个亚类,即由紫光芒等21份材料组成的第1亚类和由青海8等20份材料组成的第2亚类。主坐标分析中第1大类材料与GS聚类区分的第1大类及第2大类下的第1亚类(紫光芒除外)所属材料一致,其他GS区分的第2类(紫光芒除外)所属材料同主坐标分析的第2类材料一致。
2.3 群体结构分析

图2 基于反转录转座子标记(A)及SSR标记(B)的63份裸大麦材料聚类Fig.2 Cluster result of 63 parent materials based on retrotransposon markers (A) and SSR makers (B)
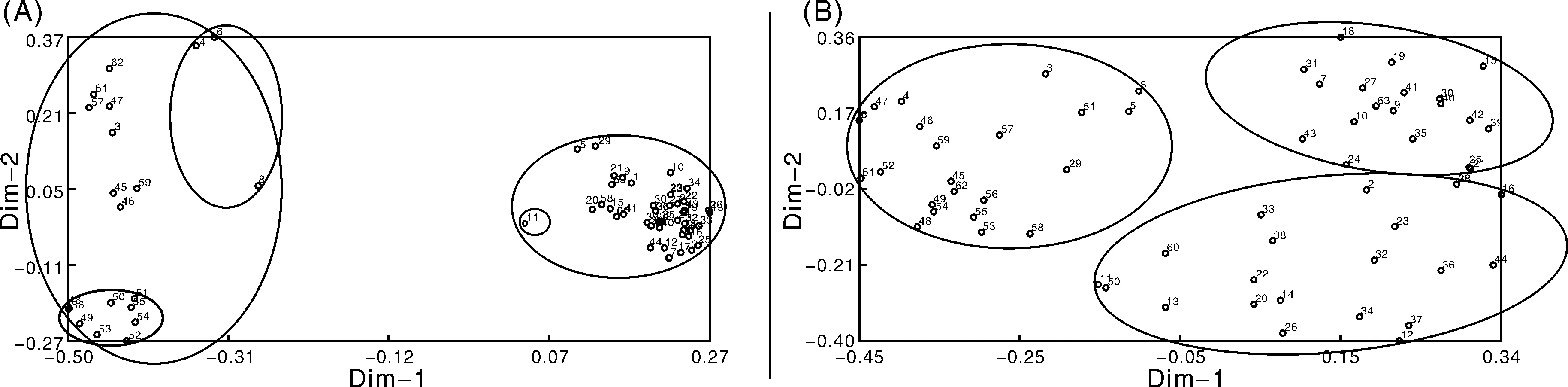
图3 63份裸大麦亲本材料反转录转座子标记(A)及SSR标记(B)主坐标分析聚类Fig.3 Principal coordinates analysis on 63 hulless barley germplasms with retrotransposon markers (A) and SSR markers (B)
基于反转录转座子标记的数据群体结构分析表明,样本的等位变异频率特征类型数K呈持续增大趋势,当K=2时,ΔK值最大,据此将63份材料分成2个亚群(图4-A),分别包含20、43份材料,其中60份材料Q值大于0.7,表明这些裸大麦材料来源单一,遗传背景简单,亚类间缺少基因交流(图5-A)。同反转录转座子标记GS聚类及主坐标分类2个类群的20、43份材料相比,分类一致。
基于SSR标记的数据群体结构分析显示,样本的等位变异频率特征类型数K呈持续增大趋势,当K=2时,ΔK值最大,据此将63份材料分成2个亚群(图4-B),分别包括23、40份材料,其中58份材料Q值大于0.7。这再次证明了这些裸大麦材料来源单一,遗传背景简单,亚类间缺少基因交流(图5-B)。但与主坐标分析、聚类分析所区分的材料大类、亚类所属类群相比,无明显重合性。
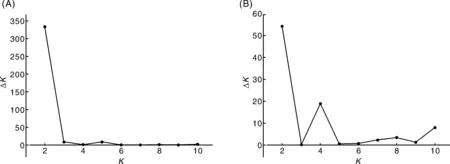
图4 基于反转录转座子标记(A)和SSR标记(B)数据的K值与ΔK折线图Fig.4 ΔK with change of K values based on retrotransposon markers (A) and SSR makers (B)

图5 基于反转录转座子标记(A)和SSR标记(B)的63份大麦材料群体遗传结构分析Fig.5 Population structure of 63 hullness barley germplasms based on retrotransposon markers (A) and SSR markers (B)
3 讨论
反转录转座子标记是鉴定和区分种质资源材料的有效方法。基于IRAP与REMAP标记可以区分葡萄牙历史上育成的所有小麦品系[15]。Biswas等[22]在柑橘中的研究也表明,反转录转座子标记是区分柑橘及其近缘种的有效手段。Kim等[23]建立了基于反转录转座子标记的日本梨品质识别方法。杜晓云等[24]利用IRAP 建立柿属植物指纹图谱,能有效区分芽变品种。肖炳光等[25]用 IRAP标记的方法对烤烟品种扩增,平均多态性比率达96%,显著优于其他测试标记分析结果。
在本研究中,IRAP、REMAP、SSR标记分别检测到315、143、38个等位变化;变异范围分别为9~58、7~25、2~4个,平均每对引物检测到24.23、14.30、2.38个。赖勇等[5,26]利用SSR标记对国内外大麦材料进行检测,检测出的等位基因变异范围为2~7个,平均每对标记检测2.9个。同这些研究相比,反转录转座子标记(IRAP、REMAP)检测到的等位基因变异范围更大,平均每个标记检测到变异数更多,说明反转录转座子水平上的遗传多样性分析能揭示更为丰富的裸大麦遗传结构信息,可为后续育种利用提供更多信息。
赖勇等[5]用SSR标记检测青稞材料的GS变幅为0.497~0.970,平均0.761;巴桑玉珍等[6]利用SSR标记分析青稞耐寒资源的GS变化范围为0.469~0.924,平均0.745;尚毅等[27]的研究表明,浙江省裸大麦地方品种遗传多样性水平较高。较以上研究,本研究揭示的裸大麦遗传相似性系数变异幅度更大,GS平均值更小,亲缘关系更远,这可能与本研究中试验材料地理来源丰富,并且使用了反转录转座子及SSR两种标记有关。
基于数学模型的群体结构分析中,本研究认为这些裸大麦材料来源单一,遗传背景简单,亚类间几乎没有基因交流,在育种利用的过程中,可以考虑加大不同类群裸大麦材料之间的交流。本研究SSR标记的群体结构分析与GS聚类结果差异较大,这可能是因为群体结构分析可以降低人为主观分类对关联分析的影响,能更准确地了解这些材料的遗传背景[28]。群体结构分析是关联作图的基础,本研究基于2种标记的群体结构分析结果存在较大差异。在后续关联分析中,应充分考虑标记类型对关联分析结果的影响。
本研究中,反转录转座子与SSR标记的GS聚类都将63份裸大麦划分为2个大的类群,但反转录转座子标记的GS聚类及主坐标分析皆能将野生祼大麦单独聚为一个小类,而SSR标记聚类结果中野生裸大麦材料未能单独聚为一类。这种差异可能是由不同标记揭示了基因组不同类型和DNA多样性所造成的。IRAP和REMAP反映基因组反转录转座子区域的结构变异,SSR标记揭示的是基因组简单重复序列的多样性。反转录转座子在植物非生物胁迫应答及基因组进化中起着重要作用[29]。已有充分的证据表明,反转录转座子驱动了大麦基因组进化,是大麦基因组响应外界胁迫的重要元件[12-14]。西藏大麦材料基因组反转录转座子区域出现了不同于其他材料的进化模式是个值得进一步研究的问题。
本研究表明,多标记协同检测可以揭示材料间更丰富的遗传背景差异。但本研究未能对不同标记间检测结果差异化的存在做更深入的探究。基于反转录转座子的标记较常规的SSR标记,提供的信息丰富,用时少,费用低[30],而且具有检测区域不同、灵敏度高、多态性好、基因组覆盖度广等诸多优势。对以上2种标记协同区分裸大麦遗传背景差异化的研究,将有利于发掘常规标记检测局限区域外有差异化的亲本材料,对多标记协同在遗传多样性分析、种质鉴定、亲缘关系分析及亲本选配中的应用提供了实例。
[1] 孟凡磊,强小林,佘奎军,等. 西藏主要农区青稞品种的遗传多样性分析[J]. 作物学报, 2007, 33(11): 1910-1914.
MENG F L, QIANG X L, SHE K J, et al. Genetic diversity analysis among hulless barley varieties from the major agricultural areas of Tibet[J].ActaAgronomicaSinica,2007,33(11):1910-1914. (in Chinese with English abstract)
[2] 杨平,刘仙俊,刘新春,等. 利用SRAP标记研究四川高原青稞育成品种的遗传多样性[J]. 遗传, 2008, 30(1): 115-122.
YANG P, LIU X J, LIU X C, et al. Genetic diversity analysis of the developed qingke (hulless barley) varieties from the plateau regions of Sichuan in China revealed by SRAP markers[J].Hereditas, 2008, 30(1): 115-122. (in Chinese with English abstract)
[3] 曾兴权,王玉林,徐齐君,等. 利用SSR引物分析西藏青稞种质资源的遗传多样性[J]. 麦类作物学报, 2013, 33(2): 260-267.
ZENG X Q, WANG Y L, XU Q J, et al. Asessment of geneic diversity in tibetan hulless barley germplasm (HordeumvulgareL.var.nudum HK.f.) by SSR primers[J].JournalofTriticeaeCrops, 2013, 33(2): 260-267. (in Chinese with English abstract)
[4] FENG Z Y, ZHANG L L, ZHANG Y Z, et al. Genetic diversity and geographical differentiation of cultivated six-rowed naked barley landraces from the Qinghai-Tibet plateau of China detected by SSR analysis[J].GeneticsandMolecularBiology, 2006, 29(2): 330-338.
[5] 赖勇,王晋民,任龙,等. 大麦SSR标记遗传多样性及连锁不平衡分析[J]. 核农学报, 2016, 30(10): 1889-1897.
LAI Y, WANG J M, REN L, et al. Genetic diversity and linkage disequilibrium analysis of barley using SSR markers[J].JournalofNuclearAgrictulturalSciences, 2016, 30(10): 1889-1897. (in Chinese with English abstract)
[6] 巴桑玉珍,刘新春,付国勇,等. 青藏高原青稞耐寒种质资源基于SSR标记的遗传多样性及群体结构分析[J]. 麦类作物学报, 2017, 37(1): 40-47.
BASANG Y Z, LIU X C,FU G Y, et al. Genetic diversity and population structure analysis of hulless barley with cold tolerance from the Qinghai-Tibetan plateau revealed by SSR markers[J].JournalofTriticeaeCrops, 2017, 37(1): 40-47. (in Chinese with English abstract)
[7] KALENDAR R, FLAVELL A J, ELLIS T H N, et al.Analysis of plant diversity with retrotransposon-based molecular markers[J].Heredity, 2011, 106(4): 520-530.
[8] TSIANTIS M. A transposon in tb1 drove maize domestication[J].NatureGenetics, 2011, 43(11): 1048-1050.
[9] WICKER T, MAYER K F X, GUNDLACH H, et al. Frequent gene movement and pseudogene evolution is common to the large and complex genomes of wheat, barley, and their relatives[J].ThePlantCell, 2011, 23(5): 1706-1718.
[10] WICKER T, YU Y, HABERER G, et al. DNA transposon activity is associated with increased mutation rates in genes of rice and other grasses[J].NatureCommunications, 2016, 7: 12790.
[11] 李书粉,李莎,邓传良,等. 转座子在植物 XY 性染色体起源与演化过程中的作用[J]. 遗传, 2015, 37(2): 157-164.
LI S F, LI S, DENG C L, et al. Role of transposons in origin and evolution of plant XY sex chromosomes[J].Hereditas, 2015, 37(2): 157-164. (in Chinese with English abstract)
[12] CAMPBELL B C, LEMARE S, PIPERIDIS G, et al. IRAP, a retrotransposon-based marker system for the detection of somaclonal variation in barley[J].MolecularBreeding, 2011, 27(2): 193-206.
[13] SHANG Y, YANG F, SCHULMAN A H, et al. Gene Deletion in barley mediated by LTR-retrotransposonBARE[J].ScientificReports, 2017, 7:43766.
[14] MASCHER M, GUNDLACH H, HIMMELBACH A, et al. A chromosome conformation capture ordered sequence of the barley genome[J].Nature, 2017, 544(7651): 427-433.
[15] CARVALHO A, GUEDES-PINTO H, MARTINS-LOPES P, et al. Genetic variability of old Portuguese bread wheat cultivars assayed by IRAP and REMAP markers[J].AnnalsofAppliedBiology, 2010, 156(3): 337-345.
[16] KALENDAR R, GROB T, REGINA M, et al. IRAP and REMAP: two new retrotransposon-based DNA fingerprinting techniques[J].TheoreticalandAppliedGenetics, 1999, 98(5): 704-711.
[17] SINGH S, NANDHA P S, SINGH J. Transposon-based genetic diversity assessment in wild and cultivated barley[J].TheCropJournal, 2017, 5(4): 296-304.
[18] 张志仙,何道根,朱长志, 等.青花菜种质资源遗传多样性的SSR分析[J]. 浙江农业学报, 2017, 29(2): 228-235.
ZHANG Z X, HE D G, ZHU C Z, et al. Genetic diversity analysis ofBrassicaoleraceaL. var.italicawith SSR markers[J].ActaAgriculturaeZhejiangensis, 2017, 29(2): 228-235. (in Chinese with English abstract)
[19] 张安世,张素敏,范定臣,等. 皂荚种质资源 SRAP 遗传多样性分析及指纹图谱的构建[J]. 浙江农业学报, 2017, 29(9): 1524-1530.
ZHANG A S, ZHANG S M, FAN D C, et al. Genetic diversity and fingerprints ofGleditsiasinensisgermplasm based on SRAP[J].ActaAgriculturaeZhejiangensis, 2017,29(9): 1524-1530. (in Chinese with English abstract)
[20] 黄孙平,陈黎,戴丽荷,等.鸡PRLR脚基因多态位点检测及其与产蛋性状的关联分析[J]. 浙江农业学报, 2015, 27(7): 1141-1147.
HUANG S P, CHEN L, DAI L H, et al. Polymorphism analysis of chicken PRLR genes and their association with layillg performances[J].ActaAgriculturaeZhejiangensis, 2015, 27(7): 1141-1147. (in Chinese with English abstract)
[21] EVANNO G, REGNAUT S, GOUDET J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study[J].MolecularEcology, 2005, 14(8): 2611-2620.
[22] BISWAS M K, BAIG M, CHENG Y J, et al. Retro-transposon based genetic similarity within the genusCitrusand its relatives[J].GeneticResourcesandCropEvolution, 2010, 57(7): 963-972.
[23] KIM H, TERAKAMI S, NISHITANI C, et al. Development of cultivar-specific DNA markers based on retrotransposon-based insertional polymorphism in Japanese pear[J].BreedingScience, 2012, 62(1): 53-62.
[24] 杜晓云,罗正荣. 部分柿属植物IRAP反应体系的建立和指纹图谱构建[J]. 农业生物技术学报, 2016, 14(6): 931-936.
DU X Y, LUO Z R.Establishment of the inter-retrotransposon amplified polymorphism (IRAP) reaction system and construction of fingerprint in someDiospyrosspp.[J].JournalofAgriculturalBiotechnology, 2016, 14(6): 931-936. (in Chinese with English abstract)
[25] 肖炳光,杨本超. 利用IRAP标记分析烤烟品种间遗传差异[J]. 西北植物学报, 2006, 26(6) : 1119-1124.
XIAO B G, YANG B C. Analysis of genetic differences among flue-cured tobacco varieties by IRAP markers[J].ActaBotanicaBoreali-OccidentaliaSinica, 2006, 26(6) : 1119-1124. (in Chinese with English abstract)
[26] 赖勇,贾建磊,王晋民,等. 外引大麦SSR标记遗传多样性及其与农艺性状的关联分析[J]. 麦类作物学报, 2017, 37(2): 197-204.
LAI Y, JIA J L, WANG J M, et al. Analysis of genetic diversity and association with agronomic traits in barley (HordeumvulgareL.) introduced from abroad using SSR markers[J].JournalofTriticeaeCrops, 2017, 37(2): 197-204. (in Chinese with English abstract)
[27] 尚毅,华为,朱靖环,等. 浙江省裸大麦地方品种遗传多样性分析[J]. 麦类作物学报, 2014, 34(7): 922-928.
SHANG Y, HUA W, ZHU J H, et al. Genetic diversity of hulless barley landraces in Zhejiang[J].JournalofTriticeaeCrops, 2014, 34(7): 922-928. (in Chinese with English abstract)
[28] GUPTA P K, RUSTGI S, KULWAL P L. Linkage disequilibrium and association studies in higher plants: present status and future prospects[J].PlantMolecularBiology, 2005, 57(4): 461-485.
[29] 王石平,张启发.高等植物基因组中的反转录转座子[J].植物学报,1998, 40(4): 291-297.
WANG S P,ZHANG Q F.Retrotranspoons in the genomes of higher plants[J].ActaBotanicaSinica, 1998, 40(4): 291-297. (in Chinese with English abstract)
[30] SMYKAL P. Development of an efficient retrotransposon-based fingerprinting method for rapid pea variety identification[J].JournalofAppliedGenetics, 2006,47(3): 221-230.
