菊苣粕果胶的微波辅助提取工艺研究
2018-03-13皮芳郭晓明刘战朋于淑娟
皮芳,郭晓明,2,刘战朋,于淑娟,2,3
(1.华南理工大学食品科学与工程学院,广东广州 510640)(2.华南理工大学广东省天然产物绿色加工与产品安全实验室,广东广州 510640)(3.华南理工大学制浆造纸工程国家重点实验室,广东广州 510640)
果胶存在于陆生植物的细胞壁及细胞间质中,是一类异质性多糖的统称[1]。从高分子化学角度上看,果胶是一种高分子共聚物[2],由聚半乳糖醛酸聚糖(Homogalacturonan,HG)、I类鼠李糖半乳糖醛酸聚糖(Rhamnogalacturonan I,RGI)、II类鼠李糖半乳糖醛酸聚糖(Rhamnogalacturonan II,RGII)等特异性的结构区域组成。这些结构区域通过复杂的方式彼此相连,因而赋予了果胶独特的结构与功能性[3,4]。
果胶是一种天然的质构改良剂,在食品工业中应用广泛。果胶的功能性质依赖于原料的种类;源于苹果渣、橘皮的果胶具有良好的凝胶性能,而来自甜菜粕中的果胶则具有理想的乳化活性[5]。近年来,随着果胶新资源的进一步挖掘,菊苣粕和百香果果皮等非传统原料逐渐受到生产者的关注[6,7]。通过筛选原料可直接获得特定结构与性能的果胶产品,既简化了加工程序,又能降低生产成本,具有重要的现实意义。因此,与新型果胶资源相关的开发与转化技术,已成为果胶生产技术的研究热点之一。
菊苣粕是菊粉加工过程产生的副产物,富含可发酵性碳水化合物[8,9]。若加工不及时,菊苣粕极容易被微生物利用,堆积时间长将导致严重的环境污染问题。据报道,菊苣粕富含果胶(12.4~24.0 wt%)类物质[10,11],被视为一种潜在的果胶原材料。菊苣源果胶具有低酯化度(<50%)的特点[7,10],可加工为低酯果胶应用于食品工业中。以菊苣粕为原料提取果胶,不但可以扩大果胶物质的来源,还可以提高菊苣粕的附加值。
然而,由于菊苣粕深加工技术落后、设备配套不完善等客观原因,我国的菊苣粕果胶产业化仍有待发展。
提取是果胶生产技术的关键单元操作之一,对于产品质量控制具有重要作用。长期以来,菊苣粕果胶的提取沿用CHE工艺[11~13]。该法以pH 1.5~2.0的稀酸溶液作为提取溶剂,在80 ℃以上的高温下萃取菊苣粕中的果胶多糖。CHE操作简单,但也存在缺陷与不足,比如耗时(1~4 h)[14]、降解果胶分子等问题[15,16]。微波是一种频率为300 MHz~300 GHz的电磁波,用于天然产物的提取,具有选择性强、操作时间短、目标组分得率高,并且能极大限度地保留分离组分的天然活性等优点[17]。针对 CHE的局限性,本文探究了菊苣粕果胶的MAE工艺,并对比了微波加热与水浴加热对果胶得率、化学组成和分子量等指标的影响,旨在为菊苣粕果胶提供一种高效的提取方法。
1 材料与方法
1.1 实验材料
菊苣粕,总糖、水分含量分别为60.4%和7.8%,由维乐夫公司提供;无水乙醇,南京化学试剂有限公司;盐酸(分析纯),北京试剂厂;果胶酶Rapidase C 80 MAX,荷兰帝斯曼公司;D-半乳糖醛酸,美国sigma公司;三苯基酚,上海梯希爱化成工业有限公司;三氟乙酸(分析纯),上海阿拉丁生化科技股份有限公司;氢氧化钠50%(色谱纯),德国Merck公司;其它试剂均为分析纯。
1.2 主要仪器设备
Mars 6微波消解仪,美国CEM公司;Fiveasy实验室pH计,梅特勒-托利多仪器有限公司;RO 10磁力搅拌器,德国IKA公司生产;GR 22高速冷冻离心机,日本日立公司;高效液相色谱 Waters 1525,Ultrahydragel Guard,Ultrahydragel 2000,Ultrahydragel 1000,美国 Waters公司;高效阴离子交换色谱ICS-5000,美国戴安公司;2414示差折光检测器,美国Waters公司;TU-1901紫外可见分光光度计,北京普析通用仪器公司;1530 VP扫描电子显微镜,德国LEO公司;Cressington 108离子溅射仪,英国Cressington公司;红外光谱仪,VERTEX 70,德国Bruker公司。
1.3 实验方法
1.3.1 菊苣粕果胶提取工艺流程

图1 微波辅助提取法(a)与传统热酸提取法(b)的提取装置示意图Fig.1 Schematic diagram of microwave extraction (a) and conventional extraction (b) devices
微波辅助提取(Microwave-assisted extraction,MAE):MAE所采用的设备简图,如图1a所示。精确称取菊苣干粕粉末1.00 g,装入Green Chem微波消解罐中,加入30 mL去离子水,随后加入180 μL、6 mol/L盐酸溶液调节pH值为2.0,静置15 min。在消解罐中加入转子(2 cm),微波提取功率为200 W,提取时间分别为45 s、90 s和120 s。提取结束,立即用冰水冷却至室温,将料液用400目滤布过滤得粗提液,倒入离心杯中,转速10000 r/min离心20 min,取上清液。将提取液缓慢加到3倍体积95%乙醇中,充分搅拌,醇沉3 h,随后以转速8000 r/min离心10 min,将絮凝物先后用75%和95%的乙醇洗涤并在相同条件下离心。将所得絮凝物以400目滤布过滤,收集体系中的絮凝物。最后置于干燥箱中以45 ℃烘至恒重,再用分析天平称重。为便于讨论,将MAE提取45 s、90 s、120 s的果胶样品分别标记为 MAE45、MAE90和MAE120。
传统热酸法(Conventional heating extraction,CHE):CHE所用设备简图,如图1b所示。参考Yapo等人的方法[6],称取菊苣干粕粉末10.0 g,装入250 mL平底烧瓶,加入250 mL去离子水,随后加入1.6 mL、6 mol/L的盐酸调节pH值为2.0,水浴温度为80 ℃,提取时间分别为0.5 h、1 h、2 h和4 h。提取结束后的分离、离心醇析、干燥等工艺同MAE。为便于讨论,将CHE提取0.5 h、1 h、2 h、4 h所得果胶样品分别标记为CHE0.5、CHE1、CHE2和CHE4。
1.3.2 菊苣粕果胶得率测定方法
果胶经分析天平称重后,用公式(1)计算果胶得率:
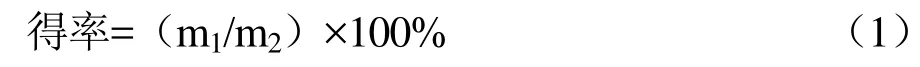
式中:m1-成品果胶质量(g);m2-菊苣粕质量(g)。
1.3.3 菊苣粕果胶理化性质的测定
1.3.3.1 半乳糖醛酸(GalA)含量测定
GalA含量测定采用紫外可见分光光度计法[18]。
GalA标准曲线:配制GalA标品溶液(50 µg/mL),分别取40、120、240、360、400 µL标品置于10 mL带塞消化管中,再相应加入360、280、160、40、0 µL去离子水,冰浴条件下加入2.5 mL浓硫酸溶液,涡旋振荡使其充分混匀后,置于100 ℃水浴中5 min使其多糖完全水解。随后加入50 µL显色剂,空白样加入50 µL 0.5% NaOH溶液,静置一段时间后,以两个空白样校零,520 nm波长下测其吸光值。
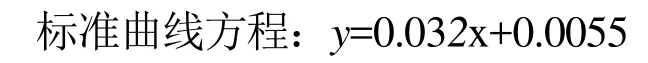
式中:y为吸光度;x为 D-半乳糖醛酸的含量(µg),R2=0.9999。
果胶样品GalA的测定:称取果胶样品5 mg充分溶解后定容到100 mL,取400 µL样品到10 mL带塞消化管中,其余步骤同上,每个样品做三次平行。
1.3.3.2 中性糖(NS)测定
NS的测定方法参考Garna等的方法[19],用高效阴离子交换色谱分析。称取10 mg果胶样品于密封消化管中,加入2 mL果胶酶溶液(E.C.3.2.1.15.,日本Amano公司),置于45 ℃水浴中24 h。再加入2 mL、4 mol/L三氟乙酸(TFA),随后120 ℃油浴1 h至完全酸解,立即将样品冰浴冷却,加7 mol/L氨水调节pH至9.0左右,用去离子水定容至100 mL。水解液过0.22 μm 滤膜后,注射入高效阴离子交换色谱(HPAEC)系统(ICS-5000,Dionex Corp.,USA)。
色谱条件:色谱柱:CarboPac PA1(4×250 mm)和CarboPac PA1保护柱(4×50 mm);检测器:电化学检测器(ED 50);流动相:100 mmol/L NaOH;流速:1 mL/min,柱温:30 ℃,进样量:25 µL;淋洗条件:0~25 min,16 mmol/L NaOH,25~35 min,500 mmol/L NaOH。数据经Chromeleon 7.2软件采集、分析后,绘制色谱曲线。
1.3.3.3 分子量测定
果胶重均分子量(Mw)的测定,利用尺寸排阻凝胶色谱(HPSEC)测定。果胶样品配制成1 mg/mL溶液,样品过 0.45 µm 滤膜后测定。色谱条件:Ultrahydragel Guard(40 mm×6 mm)、Ultrahydrogel 2000(300 mm×7.5 mm)和Ultrahydrogel 1000(300 mm×7.5 mm)串联;流动相为100 mmol/L NaNO3;流速0.6 mL/min;检测器采用示差折光检测器,柱温35 ℃;进样量 100 µL。采用葡聚糖标品(Mw=11.6~608 ku)建立分子量回归曲线,并通过 Empower软件(Version 2.0,美国Waters公司)计算Mw。
1.3.3.4 酯化度测定
甲酯化度和乙酰化度的测定采用高效液相色谱法[20]。
以异丙醇:水=1:1作为皂化液。称取25 mg果胶样品在2 mL离心管中,加入1 mL皂化液,将混合物在4 ℃下皂化3 h。皂化后以10000 r/min转速离心5 min,取上清液过0.22 μm滤膜后,用高效阴离子交换色谱测定。色谱条件:柱子:Aminex HPX-87H,美国伯乐公司;流动相:5 mmol/L H2SO4;柱温:25 ℃;流速:0.5 mL/min等度洗脱。样品平行测定2遍,数据用平均值±标准偏差表示。
1.3.3.5 衰减全反射-傅里叶变换红外光谱(ATR-FT-IR)分析
果胶的傅里叶变换红外光谱(FT-IR)分析用VERTEX 70光谱仪(德国Bruker公司)在分辨率4 cm-1时记录,区间为3700~550 cm-1。将冻干的果胶样品置于金刚石上,测定其衰减全反射(ATR)图谱。
1.3.3.6 扫描电镜(SEM)分析
将提果胶后的菊苣粕样品冻干,制成薄片,用导电双面胶固定在载物台上。样品镀金后,采用 LEO 1530 VP扫描电子显微镜观察其表面形貌。
1.4 数据分析
采用 Scheffe多重比较分析同系列样品、不同系列样品间的差异性(n=3,α=0.05);统计软件为SPSS(20.0版本)。
2 结果与讨论
2.1 提取方法及其工艺参数对菊苣粕果胶得率、化学组成和分子量的影响
2.1.1 得率
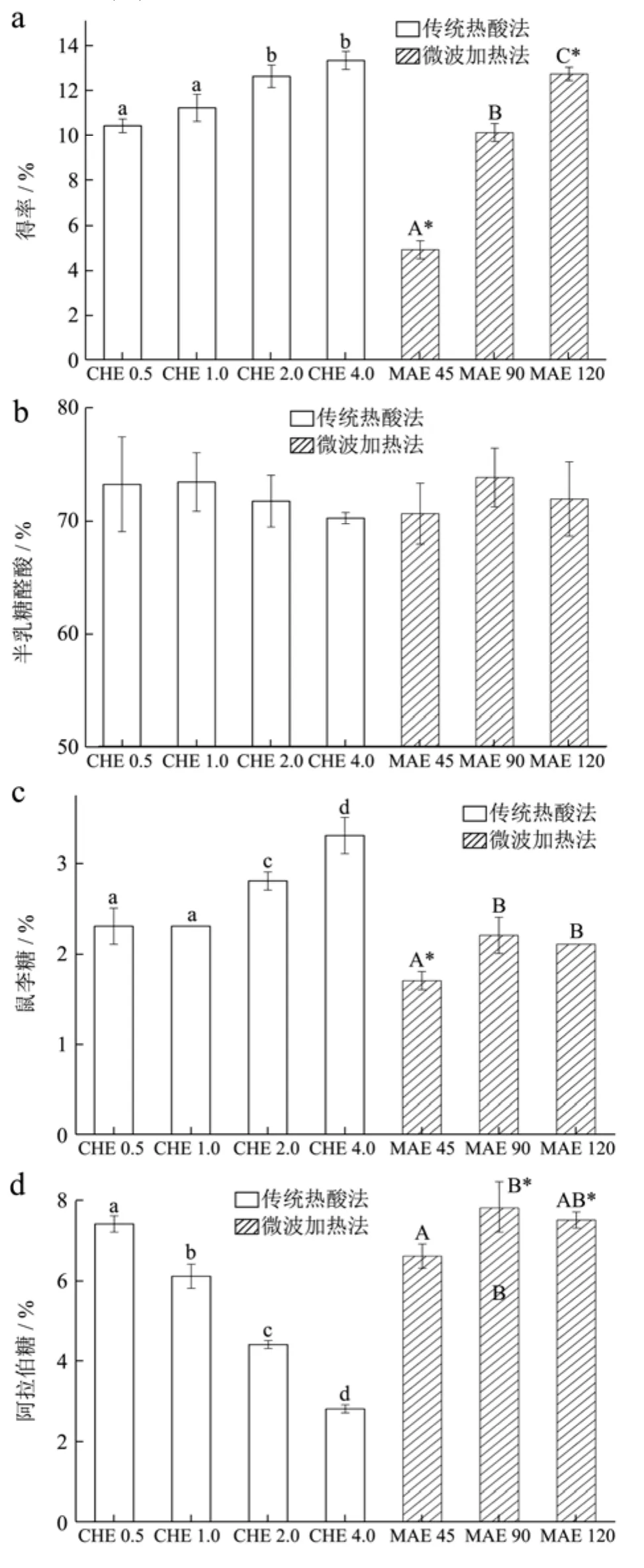

图2 两种提取方法及其工艺参数对菊苣粕果胶得率、化学组成及Mw的影响Fig.2 The effects of extraction methods and parameters on the extraction yield, structure and Mw of chicory root pulp pectin
本实验研究了两种提取方法及其工艺参数对菊苣粕果胶提取率(图2a)、化学组成(图2b~g)及Mw(图2h)的影响。如图2a所示,菊苣粕果胶的得率不仅依赖于提取方法,亦取决于受试工艺参数。采用CHE时,果胶得率随提取时间的延长而有所增加,但变化趋势并非十分显著。当提取时间从0.5 h增加到1 h时,果胶得率(10.4~11.2%)未见显著增加(p>0.05);提取时间增加到2 h后,果胶得率随之升高至12.6%;此后,尽管提取时间进一步延长至4 h,但得率基本保持不变。当采用MAE时,果胶得率随微波处理时间的延长而持续升高。微波处理45 s后,由于物料温度仅为48 ℃;在这种温和的条件下,原果胶转化为可溶性果胶的能力有限,故最终果胶得率仅为4.9%。微波处理90 s、120 s对应的物料温度为76 ℃、93 ℃,在热酸作用下,原果胶迅速转化为可溶性果胶,故果胶得率也迅速上升至10.1%、12.7%。
单因素方差分析结果表明,CHE1与MAE90两者的果胶得率无显著性差异(p>0.05),从而间接说明水浴80 ℃加热1 h与微波处理90 s具有相同的提取效果。同理,由于MAE120与CHE2在果胶得率上无显著差异,故微波处理120 s也能达到80 ℃下加热2 h的提取效果。
2.1.2 半乳糖醛酸
如图 2b所示,CHE样品的 GalA含量介于70.2~73.2%间,各样品间未见显著性差异(p>0.05)。Robert等[21]研究了提取时间对菊苣粕果胶GalA含量的影响,他们也未发现提取时间与GalA含量间存在关联性。另一方面,MAE样品的 GalA含量为70.6~73.8%,各样品间也不存在显著性差异(p>0.05)。
2.1.3 鼠李糖
鼠李糖(Rha)是果胶RGI结构区域的组成单元之一,其通过α-1,4-糖苷键与GalA交替相连[22]。如图2c所示,当提取时间不超过1 h时,CHE样品的Rha含量为2.3%;当延长提取时间至4 h过程中,CHE样品的Rha含量显著升高(3.3%)。Thibault等[16]研究了Rha-GalA糖苷键在热酸条件下(pH 1.5,80 ℃)的水解特性,发现该糖苷键具有较高的稳定性,在相同的水解时间内,Rha的保留率较高,所以Rha含量不论在CHE还是MAE条件下,都相对较稳定。MAE45的Rha含量显著低于其他MAE样品。此外,MAE45的Rha含量也低于CHE1(p<0.05)。必须指出,MAE45可能含有部分非果胶物质,其Rha含量可能受其他杂质的干扰。
2.1.4 阿拉伯糖
如图2d所示,CHE样品的阿拉伯糖(Ara)含量为2.8~7.4%。随提取时间的延长,Ara含量呈直线下降的趋势。研究表明,果胶侧链上的Ara多以阿拉伯聚糖的形式存在[23],且阿拉伯呋喃糖之间的糖苷键是果胶中性糖的连接键中最不耐酸的[16]。Guo等[24]在研究甜菜果胶的提取工艺时发现,在pH 1.5、提取温度为85 ℃的条件下,将提取时间2 h延长至3 h后,阿拉伯糖含量由2%下降至0.8%,下降幅度高达60%。在 CHE的强酸作用下,随提取时间的延长,阿拉伯聚糖侧链水解程度加剧,而总的NS含量没有显著性变化(p>0.05),所以耐酸性比较强的Rha和Gal含量则有相对提高。Ara(6.6~7.8%)是MAE样品中含量最高的中性糖,随微波处理时间的增加,其含量并未降低。与之相反,当微波处理时间由45 s增至90 s时,Ara含量还有所升高。
可见,本文所采用的MAE参数较为温和,有效避免了Ara单元的降解。由于MAE样品遭受的微波处理时间很短(120 s以内),这种短时处理方式也有助于防止阿拉伯聚糖水解的发生。与 CHE1相比,MAE90、MAE120的Ara含量更高,表明Ara单元在MAE过程中发生的降解程度更低。
2.1.5 半乳糖
如图2e所示,CHE样品的半乳糖(Gal)含量随提取时间的延长而显著升高,并且Gal(7.0~12.3%)是CHE样品中含量最高的中性糖。以CHE4、CHE0.5为例,前者的 Gal含量(12.3%)是后者(7.0%)的1.8倍,这说明Gal在提取过程中得到富集。Gal不仅来源于果胶的中性糖侧链,还可能来自外源性半乳聚糖。研究结果表明,CHE在提取果胶的同时,也不可避免引入了部分非果胶多糖[25]。结合本文的结果,Gal含量随提取时间递增的趋势,很可能是半乳聚糖等杂多糖的含量逐渐升高所致。MAE样品的Gal含量介于4.8~5.6%,显著低于CHE1(p<0.05)。对于MAE样品而言,当微波时间由90 s升高至120 s时,Gal含量也表现出升高的趋势,但上升的幅度较小。
2.1.6 木糖
如图 2f所示,CHE样品的木糖(Xyl)含量为0.5~0.6%,MAE样品的Xyl含量为0.4~0.6%。单因素分析结果表明,提取方法、提取条件均未对Xyl含量产生显著性的影响(p>0.05)。
2.1.7 中性糖
两种提取方法得到的菊苣粕果胶的NS的单糖组成类型一致,主要成分均为Rha、Ara和Gal,其NS含量均比甜菜粕果胶的NS含量低[14]。如图2g所示,CHE样品的中性糖含量为17.2~19.2%。尽管Ara和Gal含量存在显著差异(p<0.05),但CHE样品的NS含量未见显著性的差异(p>0.05),这是由于各种单糖加和成NS后,不同单糖间的差异性被抵消。另一方面,MAE样品的NS含量为13.8~15.6%,各样品间不存在显著性差异(p>0.05),这说明微波处理时间不是决定NS含量的关键参数。
2.1.8 分子量

图3 CHE 样品(a)、MAE 样品(b)的分子量分布曲线的影响Fig.3 Molecular weight distribution profiles of the CHE (a) and MAE (b) samples
如图2h所示,CHE样品的Mw大小与提取时间具有很强的相关性。在较短的提取时间内(不超过 1 h),尽管菊苣粕果胶Mw在276~283 ku内轻微波动,但Mw未发生显著性变化(p>0.05)。随着提取时间的进一步增加,CHE样品的Mw却逐渐降低显著减小。在提取时间为4 h时,CHE4的分子量降低至228 ku,CHE样品的分子量分布曲线如图3a所示。
不同样品的分子量曲线很相似,具有宽分布、多分散的特点,这也是果胶分子量分布曲线的典型特征之一[26]。CHE样品由两个不同尺寸的组分构成,分别对应分子量分布曲线21~27 min、27~36 min内淋洗出的物质。
通过比较不同 CHE样品间的分子量分布曲线,可以发现以下规律:提取时间越长,大分子组分含量逐渐减少,而小分子组分含量则逐渐增加。以上结果表明,长时间的热酸作用导致菊苣粕果胶分子结构发生显著的降解。
相反,MAE样品的Mw却随着处理时间的增加而逐渐升高。此外,MAE样品在Mw(图2h)及果胶得率上(图2a)的变化趋势高度一致。
为了进一步阐述导致菊苣粕果胶Mw在微波场中的变化原因,本文比较了CHE、MAE系列样品的分子量分布曲线。如图3b所示,MAE45样品的分子量分布曲线为单一的宽分散曲线,且具有良好的对称性。但与其他 MAE样品相比,MAE45中大分子组分(20~28 min)的含量很低。然而,随着微波处理时间的延长,大分子组分的相对含量依次增加。综合Mw与得率的变化规律,菊苣粕果胶在微波场中的提取过程可能分为两个阶段:(1)MAE初期(≤45 s),主要是小分子果胶片段从菊苣粕中溶出;(2)MAE中期(45~120 s),主要是大分子果胶片段从菊苣粕中溶出。
2.1.9 甲酯化度与乙酰化度
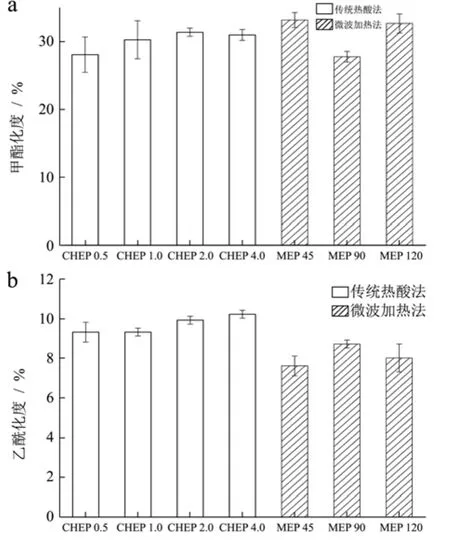
图4 提取方法及其工艺参数对甲酯化度(a)、乙酰化度(b)的影响Fig.4 Effects of extraction methods and parameters on degrees of metylation (a) and acetylation (b)
如图4所示,CHE样品的甲酯化度、乙酰化度分别为28.0~31.3%、9.3~10.2%;MAE样品的甲酯化度、乙酰化度分别为27.7~33.1%、7.6~8.7%;这些数据与Robert等[21]报道的甲酯化度(21.0~57.0%)、乙酰化度(5.0~16.0%)接近。此外,尽管提取方法、处理时间不同,但所有产品均为低酯果胶。
2.2 物料温度随时间的变化

图5 物料温度随微波处理时间(a)、水浴对流加热时间(b)的变化Fig.5 Temperature changes of the extraction medium as a function of microwave radiation time (a) and convective heating time (b), respectively
如图5所示,在微波场(200 W)中,物料吸收微波后,导致物料温度呈线性攀升。微波具有很高的加热效率,处理120 s后,物料温度从22 ℃升高至93 ℃(图 5a)。
相反,以外部热水作为热源时,物料温度升高至77 ℃耗时13.5 min(图5b);随着物料与外部热源的温度差的逐步减小,物料升温速率逐渐变缓,达到目标温度(80 ℃)总耗时为30 min。
物料温度是影响果胶得率、果胶分子结构的关键因素之一。由于两种提取方法对物料的加热时间、加热效率不同,其对菊苣粕果胶的得率、化学组成和分子量等指标均产生一定影响。
2.3 FT-IR-ATR
如图 6所示,菊苣粕果胶在 3700~2800 cm-1、1800~1500 cm-1内具有典型的果胶光谱特征。3384 cm-1处的吸收峰对应于OH-的拉伸振动[27];2936 cm-1处的吸收峰源于 C-H的伸缩振动;1735、1608 cm-1分别对C=O与-COO-1的拉伸振动[28]。
不同样品的FT-IR光谱图极为相似,表明两种提取方式及其工艺参数对菊苣粕果胶的主要官能团没有显著的影响。

图6 菊苣粕果胶的红外光谱分析Fig.6 FT-IR spectra of pectin in chicory root pulp
2.4 不同处理条件对菊苣粕表面形貌的影响

图7 不同方式处理的菊苣粕的SEM图Fig.7 SEM diagrams of chicory root pulp with different treatments
不同方式处理的菊苣粕的SEM图如图7所示,其放大倍数均为200倍。不同干燥方式对菊苣粕的表面形貌具有显著的影响。经热风干燥处理(图 7a),菊苣粕质地致密,但大小不均一,其表面粗糙、高低不平,由不规则的褶皱组成。经冷冻干燥处理(图7b),菊苣粕表面由大小不一的孔洞组成,呈“蜂巢”状,质地疏松。在冻干过程中,菊苣粕内部中的水分经升华除去。因此,菊苣粕表面的孔洞可能是由于水分升华后形成的“通道”。经热酸处理后(图 7c),菊苣粕质地变得更加疏松,其表面也由“蜂巢”状转变为无规则的碎片状。经 MAE120处理后的菊苣粕的形貌与CHE2处理的效果类似(图 7d)。以上结果表明,酸协同温度导致菊苣粕的形貌、结构发生剧变,为果胶的释放创造了良好的条件;微波处理120 s能达到与传统热酸法2 h相同的效果。
3 结论
3.1 本文研究了菊苣粕果胶的MAE工艺,并比较了MAE与CHE对菊苣粕果胶得率、化学组成和分子量的影响。研究结果表明,微波使物料迅速受热,从而加速了菊苣粕果胶的提取过程。与CHE相比,MAE能显著提高菊苣粕果胶的提取效率,同时降低菊苣粕果胶分子的降解程度。
3.2 从果胶得率、菊苣粕形貌和果胶分子结构等方面综合比较,微波处理120 s能达到等同于水浴80 ℃加热2 h的提取效果。
[1]D J Cosgrove. Growth of the plant cell wall [J]. Nature Reviews Molecular Cell Biology, 2005, 6(11): 850-861
[2]J Chen, W Liu, C M Liu, et al. Pectin modifications: a review[J]. Critical Reviews in Food Science and Nutrition, 2015,55(12): 1684-1698
[3]S Christiaens, S V Buggenhout, K Houben, et al.Process-structure-function relations of pectin in food [J].Critical Reviews in Food Science and Nutrition, 2016, 56(6):1021-1042
[4]B M Yapo, D Gnakri. Pectic polysaccharides and their functional properties [J]. Polysaccharides: Bioactivity and Biotechnology, 2015, 6(4): 1729-1749
[5]E D Ngouémazong, S Christiaens, A Shpigelman, et al. The emulsifying and emulsion-stabilizing properties of pectin: a review [J]. Comprehensive Reviews in Food Science and Food Safety, 2015, 14(6): 705-718
[6]B M Yapo, K L Koffi. Yellow passion fruit rind-a potential source of low-methoxyl pectin [J]. Journal of Agricultural and Food Chemistry, 2006, 54(7): 2738-2744
[7]M Panouillé, J F Thibault, E Bonnin. Cellulase and protease preparations can extract pectins from various plant byproducts [J]. Journal of Agricultural and Food Chemistry,2006, 54(23): 8926-8935
[8]U S Ramasamy, H A Schols, H Gruppen. Characteristics of bacterial enzymes present during in vitro fermentation of chicory root pulp by human faecal microbiota [J]. Bioactive Carbohydrates and Dietary Fibre, 2014, 4(2): 115-124
[9]U S Ramasamy, K Venema, H A Schols, et al. Effect of soluble and insoluble fibers within the in vitro fermentation of chicory root pulp by human gut bacteria [J]. Journal of Agricultural and Food Chemistry, 2014, 62(28): 6794-6802
[10]潘润全.菊苣果胶的提取工艺及其性质研究[D].广州:华南理工大学,2014 PAN Run-quan. Extraction and physicochemical characterization of chicory root pulp pectin [D]. Guangzhou:South China University of Technology, 2014
[11]U S Ramasamy, H Gruppen, H A Schols, Structural and water-holding characteristics of untreated and ensiled chicory root pulp [J]. Journal of Agricultural and Food Chemistry,2013, 61(25): 6077-6085
[12]C Robert, T H Emaga, B Wathelet, et al. Effect of variety and harvest date on pectin extracted from chicory roots(Cichorium intybus L.) [J]. Food Chemistry, 2008, 108(3):1008-1018
[13]H Garna, N Mabon, K Nott, et al. Kinetic of the hydrolysis of pectin galacturonic acid chains and quantification by ionic chromatography [J]. Food Chemistry, 2006, 96(3): 477-484
[14]B M Yapo, C Robert, I Etienne, et al. Effect of extraction conditions on the yield, purity and surface properties of sugar beet pulp pectin extracts [J]. Food Chemistry, 2007, 100(4):1356-1364
[15]Y J Cho, J K Hwang. Modeling the yield and intrinsic viscosity of pectin in acidic solubilization of apple pomace [J].Journal of Food Engineering, 2000, 44(2): 85-89
[16]J F Thibault, C M G C Renard, M A V Axelos, et al. Studies of the length of homogalacturonic regions in pectins by acid hydrolysis [J]. Carbohydrate Research, 1993, 238(93): 271-286
[17]J P Maran, V Sivakumar, K Thirugnanasambandham, et al.Microwave assisted extraction of pectin from waste Citrullus lanatus fruit rinds [J]. Carbohydrate Polymers, 2014, 101(2):786-791
[18]N Blumenkrantz, G Asboe-Hansen. New method for quantitative determination of uronic acids [J]. Analytical Biochemistry, 1973, 54(2): 484-489
[19]H Garna, N Mabon, B Wathelet, et al. New method for a two-step hydrolysis and chromatographic analysis of pectin neutral sugar chains [J]. Journal of Agricultural and Food Chemistry, 2004, 52(15): 4652-4659
[20]A G J Voragen, H A Schols, W Pilnik. Determination of the degree of methylation and acetylation of pectins by HPLC [J].Food Hydrocolloids, 1986, 1(1): 65-70
[21]C Robert, T Devillers, B Wathelet, et al. Use of a Plackett-Burman experimental design to examine the impact of extraction parameters on yields and compositions of pectins extracted from chicory roots (Chicorium intybus L.)[J]. Journal of Agricultural and Food Chemistry, 2006, 54(19):7167-7174
[22]A G J Voragen, G J Coenen, R P Verhoef, et al. Pectin, a versatile polysaccharide present in plant cell walls [J].Structural Chemistry, 2009, 20(2): 263-275
[23]A Nakamura, H Furuta, H Maeda, et al. Structural studies by stepwise enzymatic degradation of the main backbone of soybean soluble polysaccharides consisting of galacturonan and rhamnogalacturonan [J]. Bioscience, Biotechnology, and Biochemistry, 2002, 66(6): 1301-1313
[24]X Guo, X Guo, H Meng, et al. Using the high temperature resistant pH electrode to auxiliarily study the sugar beet pectin extraction under different extraction conditions [J].Food Hydrocolloids, 2017, 70: 105-113
[25]B M Yapo. Pectin quantity, composition and physicochemical behaviour as influenced by the purification process [J]. Food Research International, 2009, 42(8): 1197-1202
[26]M L Fishman, D T Gillespie, S M Sondney, et al. Intrinsic viscosity and molecular weight of pectin components [J].Carbohydrate Research, 1991, 215(1): 91-104
[27]A A Kamnev, M Colina, J Rodriguez, et al. Comparative spectroscopic characterization of different pectins and their sources [J]. Food Hydrocolloids, 1998, 12(3): 263-271
[28]R Gnanasambandam, A Proctor. Determination of pectin degree of esterification by diffuse reflectance Fourier transform infrared spectroscopy [J]. Food Chemistry, 2000,68(3): 327-332
