Dual-specificity phosphatase 6 (DUSP6): a review of its molecular characteristics and clinical relevance in cancer
2018-03-08MuhammadKhairiAhmadNurAininaAbdollahNurulHusnaShafieNarazahMohdYusofSitiRazilaAbdulRazakOncologicalandRadiologicalSciencesClusterAdvancedMedicalandDentalInstituteUniversitiSainsMalaysiaKepalaBatasPulauPinang00MalaysiaDepa
Muhammad Khairi Ahmad, Nur Ainina Abdollah, Nurul Husna Shafie, Narazah Mohd Yusof, Siti Razila Abdul RazakOncological and Radiological Sciences Cluster, Advanced Medical and Dental Institute, Universiti Sains Malaysia, Kepala Batas, Pulau Pinang 00, Malaysia; Department of Nutrition and Dietetics, Faculty of Medicine and Health Sciences,Universiti Putra Malaysia, Jalan UPM, Serdang, Selangor 4400, Malaysia; Regenerative Medicine Cluster, Advanced Medical and Dental Institute, Universiti Sains Malaysia, Kepala Batas, Pulau Pinang 00, Malaysia
Introduction
Overview of MAPKs
The signaling pathway of mitogen-activated protein kinases(MAPKs), which are also called extracellular signal-regulated protein kinases (ERKs), is a highly conserved signal transduction pathway with many important roles. The MAPK pathway is involved in modulating diverse cellular and physiological processes, such as cell growth, survival,apoptosis, inflammation, proliferation, differentiation,migration, development, immunity, and stress responses1-6.It is also associated with human diseases, such as obesity,diabetes, rheumatoid arthritis, neurodegenerative disorders,and cancer7-9. The MAPK pathway responds to extracellular stimulation by Gi-coupled receptor (GPCR), ultraviolet irradiation, genotoxic agents, and oxidative stress, and it also can be activated by growth factors and inflammatory and cytokine stimulation10.
MAPKs are proline-directed kinases that phosphorylate sites containing serine/threonine and tyrosine residues. They are activated by dual specificity MAPK kinases (MAPKKs),which are themselves activated by MAPKK kinases(MAPKKKs)11. The MAPKs at the end of these signaling cascades phosphorylate their target proteins, including transcription factors, translational regulators, MAPK-activated protein kinases (MAPKAP kinases), phosphatases,and other classes of proteins12. Thus, they regulate multiple biological activities, including cell proliferation,differentiation, cell cycle progression, and survival13.
The most intensely studied members of the MAPK pathway include ERK1/2 and ERK 5, the p38 kinases (p38a,b, g, and d), and c-Jun N-terminal kinase (JNK) 1, 2, and 314.Activation of an ERK signaling pathway plays a role in mediating cell division, migration, and survival. ERK1 and ERK2 are activated by MAPK/ERK kinase (MEK) 1 and MEK215. The p38 kinases are activated by dual MAP kinase kinases (MKKs) termed MKK3 and MKK6. Activation of p38 signal transduction plays an important role in the regulation of inflammation, cell cycle, cell death, development, cell differentiation, senescence, and tumorigenesis16,17. JNK,which is also known as stress-activated protein kinase(SAPK), is activated by the upstream MKK4 and MKK7 kinases via dual phosphorylation of the Thr-Pro-Tyr motif18.Activation of JNK thereby mediates the regulation of transcription factors, such as c-Jun, c-Fos, activating transcription factor 2 (ATF-2), activator protein 1 (AP-1),p53, and Elk, and also phosphorylates many cytoplasmic substrates, such as cytoskeletal proteins and mitochondrial proteins like Bcl-2 and Bcl-xl19,20. Subsequently, a plethora of cellular processes are triggered, including cell proliferation,apoptosis, autophagy, motility, metabolism, and DNA repair21, as reviewed by Sui et al.10.
MAPK signaling pathway in cancer
Aberrant regulation of MAPK cascades contributes to the development of many pathological conditions, including renal cell carcinoma22, pancreatic cancer23, bladder cancer24,and leukemia25. Regulation of these cellular processes is complex and involves external growth factors and components, such as JNK, ERKs, and the p38 groups26. The JNK signal transduction pathway is associated with the transformation of many growth factor-mediated pathways and oncogenes, such as c-Jun, ATF2, p53, NFAT4, Elk1, and DPC26. Dysregulation of the JNK pathway affects survival signaling and apoptosis in mammalian cells. ERK1 and 2 are involved in the ERK pathway, which is considered to be the best characterized MAPK signaling pathway.
Activation of ERK1 and 2 leads to transactivation and causes changes in the expression of Ets, Elk1, Myc, and estrogen receptor (ER), which are important in cell differentiation, growth, and mitosis26. Activation of JNK and ERK in association with connexin43 (Cx-43) might be involved in promoting bladder tumorigenesis via targeting the MAPK pathway24. In prostate cancer, activation of ERK1/2 by osteopontin (OPN) through c-Raf and MEK1/2 promotes cell cycle arrest in PC3 human prostate cancer cells27. The oncogenesis activity of Raf/MEK and ERK in promoting cell cycle arrest in prostate cancer cells may be regulated by p53 restoration28,29. Introduction of the wild type p53 into p53 knockout cell lines, such as PC3 and DU145, activates and increases the expression of the RAF/MEK/ERK cascade and improves its sensitivity to chemotherapeutic drugs28,29.
The p38 pathway is another well-studied component of the MAPK pathway. It is activated by cellular stress, including heat shock, high osmotic stress, protein synthesis inhibitors,proinflammatory cytokines, and certain mitogens26.Activation of the p38 MAPK pathway is essential in determination of cell fate, differentiation, and progression of somatic cells, but not during the G2/M transition and mitosis30,31. The activated p38 MAPK signaling pathway regulates various proteins related to Gaucher’s disease,inflammatory bowel disease, glioblastoma multiforme, and several cancers, such as pancreatic, lung, and breast cancer23,32-36.
MAPKs are reported to have a significant relationship with various transcription factors, such as reactive oxygen species(ROS)37, epidermal growth factor receptor (EGFR)23, Cx4324,and others. In somatic cells, ROS-mediated MAPK activation regulates cellular processes, such as cell proliferation, growth,survival, and death26,38. The accumulation of ROS is essential for prolonged MAPK activation and cell death38. Moreover,aberrant activation of EGFR/p38-MAPK signaling has been shown to control angiogenesis in pancreatic ductal adenocarcinoma by regulating the expression of Sp1-induced cyclooxygenase-2 and vascular endothelial growth factor23.
Better understanding of the MAPK network will lead to new strategies to design a system to deliver anticancer drugs to cancer cells. Establishment of positive or negative regulatory feedback loop is important to discovering new ways of looking at the role of the MAPK pathway in cancer treatment. Activation of MAPK and its molecules in combination with anticancer drugs can significantly induce apoptosis in PC-3 human prostate cancer37, colon cancer39,and leukemia25cells. Recent reports have shown that anticancer drugs, such as Toyocamycin, can reduce cell viability and enhance apoptosis in human prostate cancer cells via crosstalk between ROS and the p38/ERK MAPK signaling pathway37. The involvement of p38 activation in inducing apoptosis is in accordance with studies of diosgenin, which is a chemotherapeutic agent used to treat colon cancer39. Understanding of MAPK activities with transcription factors and components in cancer cells with or without anticancer drugs is critical to understanding the involvement of these proteins in cancer regulation.
In this review, we focus on the dual specificity phosphatases (DUSPs), particularly DUSP6. DUSPs negatively regulate MAPK members (MAPK/ERK,SAPK/JNK, and p38) that are associated with cellular proliferation and differentiation40. This review demonstrates our depth of understanding of DUSP6 as a natural terminator of MAPK function and regulation, and thereby as a pharmacological inhibitor in cancer disease. Better understanding of how DUSP6 works in carcinogenesis and in the MAPK signaling pathways may provide insights into improved diagnosis, treatment, and prognosis of cancer.
MAPKs and DUSPs
DUSPs and MAP kinase phosphatases (MKPs) have been recognized as key players in the negative regulation of MAPKs by dephosphorylating either the threonine or tyrosine residue, or both, of a conserved signature T-Y-X motif within the activation loop of the kinase41-43. Ten distinct subfamilies of catalytically active MKPs within the larger family of DUSPs have been characterized from mammalian genomes41. They are subcategorized into three subgroups based on sequence homology, subcellular localization, and substrate specificity (Table 1)44. Class I consists of four inducible nuclear MKPs (DUSP1/MKP-1,DUSP2, DUSP4/MKP-2, and DUSP5). Class II consists of three ERK-specific MKPs (DUSP6/MKP-3, DUSP7/MKP-X,and DUSP9/MKP-4), and Class III contains three MKPs that inactivate p38 and stress-activated JNK MAPKs (DUSP8,DUSP10/MKP-5, and DUSP16/MKP-7).
DUSP6
DUSP6 gene organization and evolution
The subcategorization of DUSPs into three subgroups is also supported by the phylogenetic tree of DUSP sequences with consideration of substrate preferences45(Figure 1). Class II may have diverged earlier, as the central protein is encoded by a single exon41. Class I and III may be more closely related to each other, as the central portion of the protein is encoded by two exons41.

Figure 1 Phylogenetic tree of DUSP sequences. Vector NTI software was used to derive the phylogenetic tree of the mouse DUSP amino acid sequence alignment45. Sequence differences between proteins in each DUSP protein are proportionate to the length of the branches of the phylogenetic tree.
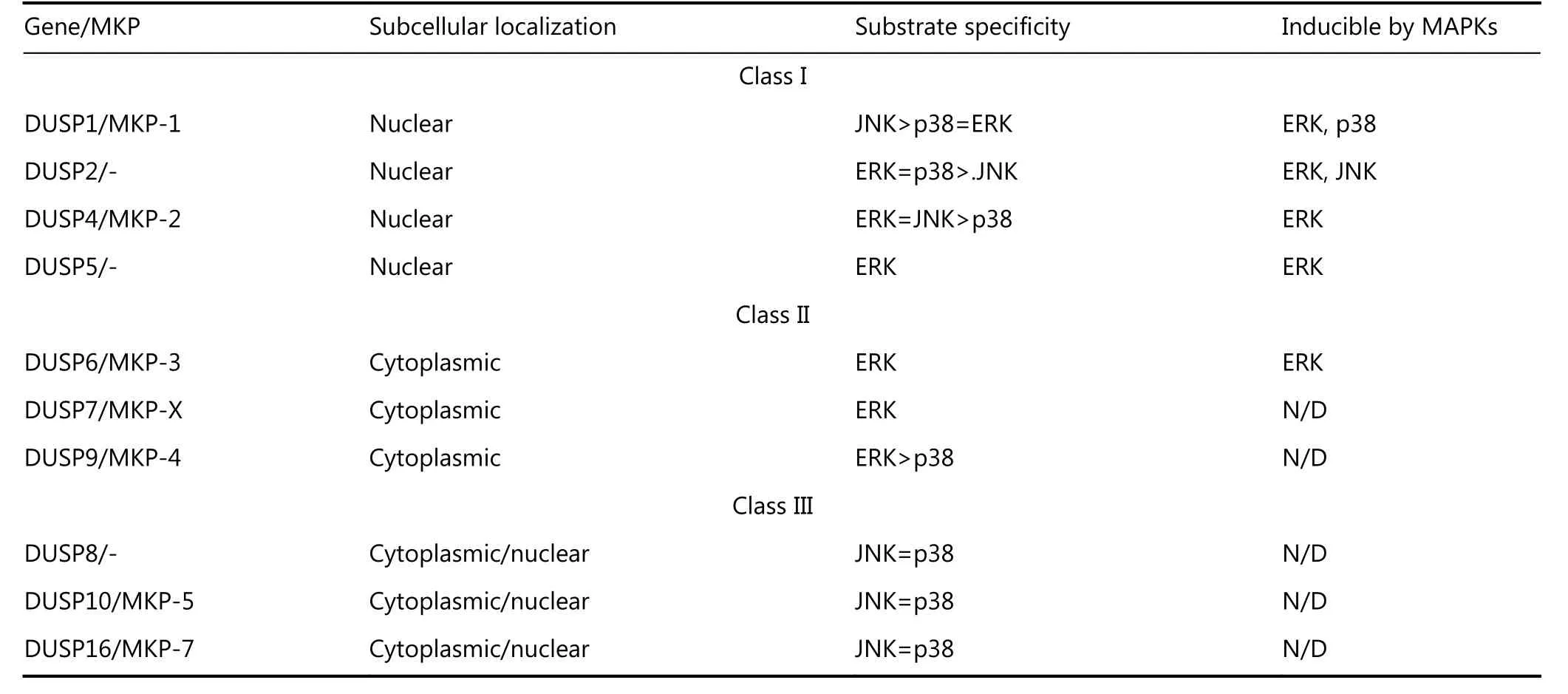
Table 1 Classification of DUSP genes in mammalian genomes into three subgroups (I, II, or III) based on sequence homology, subcellular localization, and substrate specificity41,44.
All MKPs share a common N-terminal non-catalytic domain and a more conserved C-terminal catalytic domain.The C-terminal catalytic domain consists of an active site sequence that displays sequence similarity to the prototypic dual-specificity protein phosphatase VH-1 encoded by vaccinia virus46. The non-catalytic domain consists of two regions with sequence similarity to the catalytic domain of Cdc25 cell cycle regulatory phosphatase46. These findings illustrate that the domain of MKPs and Cdc25 share a common evolutionary origin with the Rhodanese family of sulphotransferases46,47. In addition, the non-catalytic domain contains sequences that play a role in determining subcellular localization, as it harbors either a nuclear localization signal(NLS) or a nuclear export signal (NES)48,49. DUSP6 contains a leucine-rich NES that is important for nuclear export of the phosphatase50. All Class II MKPs/DUSPs, including DUSP6,share similar domain structure position1. The non-catalytic domain also contains a conserved cluster of basic amino acid residues, known as the kinase interaction motif (KIM), which is involved in MAPK substrate recognition and binding44,48,49.
Human DUSP6 is located on chromosome 12q21.33 and contains three exons consisting of 381 amino acids. The first exon encodes for the Cdc25/rhodanese-homology domain,KIM, and ends with the second Cdc25/rhodanese-homology domain41. Half of exon two and almost all sequences of exon 3 encode the functional phosphatase domain. The catalytic site is located in the third exon (Figure 2).
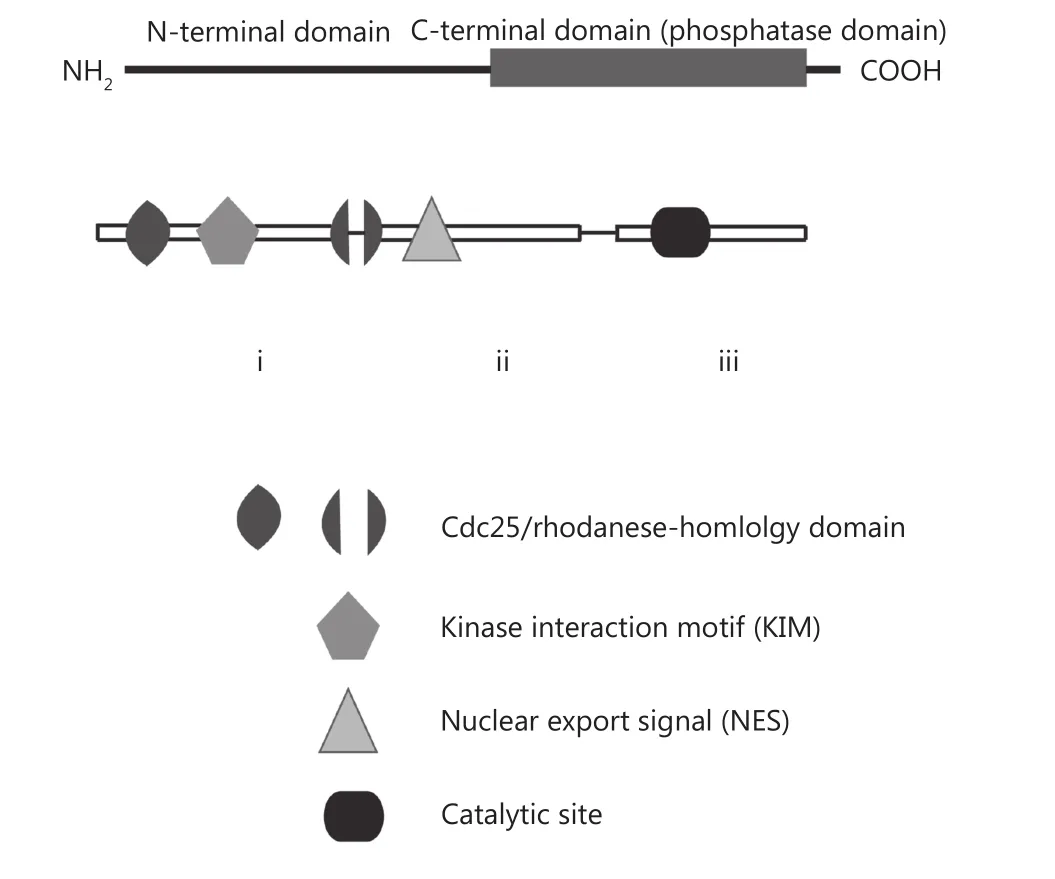
Figure 2 The domain structure of MKP-3/DUSP6 is made up of the C-terminal catalytic domain and N-terminal non-catalytic domain consisting of the Cdc25/rhodanese-homology domain,kinase interaction motif (KIM), and nuclear export signal (NES).Domains in the encoded proteins are indicated by the shaded shapes, and the three exons of DUSP6 denoted by roman numerals (rectangles) are connected through lines representing introns41,44,50,51.
Structural elements of DUSP6
The C-terminal domain contains a highly conserved protein tyrosine phosphatase (PTPase), DX26 (V/L)X(V/I)HCXAG(I/V)-SRSXT(I/V)XXAY(L/I)M, where X is any amino acid52. In DUSP6, the highly conserved C-terminal domain is made up of a tyrosine/threonine specific phosphatase designated sequence HCXXXXXR at the active site53. The cysteine plays an important role in the nucleophilic attack of the phosphorus of the substrate ERK2,whereas arginine interacts directly with the phosphate group on phosphotyrosine or phosphothreonine for transition-state stabilization53,54. This leads to rearrangement of the active residues, which enables the dephosphorylation of Thr183 and Tyr185 in ERK255. Thus, DUSP6 must undergo conformational rearrangement for its activation.
The KIM sequence in the N-terminal domain is conserved in all MKP/DUSP species56. KIM has great substrate selectivity. It mediates the binding of MKPs/DUSPs with the conserved MAPK common docking domain without requiring the phosphorylation of MAPK for activation41. The KIMDUSP6docking site consists of negatively charged, highly acidic residues and a hydrophobic groove between the α and β reverse turns of the structure56. It is located in a noncatalytic region opposite the kinase catalytic pocket56. This leads to the binding of negatively charged residues on MKPs/DUSPs to the positively charged MAPK docking site,suggesting that the MKP/DUSP binding site is critical in determining the binding specificity and catalytic activity57.Binding of specific MAPK substrates activates the catalytic activity by causing an allosteric rearrangement of the C-terminal catalytic domain, thus mediating the enzymesubstrate interaction55,58.
DUSP6 regulation and interaction in cells
DUSP6 is tightly regulated at transcription, aftertranscription, or after-translation59-63. Transcriptional regulation occurs in the presence of putative regulatory sequences for forkhead transcription factors, including Etsfamily transcription factors and phosphatidylinositol 3-kinase (PI3K) in the mTOR pathway64. Functional analysis of the promoter regions of DUSP6 has demonstrated the involvement of Ets-family transcription factors in controlling the expression of DUSP6 in chick embryo after being activated by FGF8 signaling64-66. The involvement of Etsfamily transcription factors has functional impacts through the ERK pathway, especially via a negative feedback loop of FGF signaling. A luciferase reporter assay showed that the transcriptional activation of DUSP6 by FGF family members was dependent on intron 1 in the phosphatase gene. In addition, other ERK-inducing agonists play roles in the DUSP6 transcription regulation, such as EGF in Drosophila and PDGF62,67.
Post-transcription regulation of DUSP6 can occur in several ways, which determine the stability of DUSP6 mRNA.MEK/ERK signaling and hypoxia have been demonstrated to regulate post-transcription of DUSP6. The half-life of DUSP6 mRNA in a 3’-untranslated region (3’-UTR)-dependent manner was shown to be increased by the luciferase reporter construct, Tk-luc, after Raf: ER was stimulated with 4-OHT768. Hypoxia as a result of basal ERK activity was found to increase the endogenous level of DUSP6 mRNA and the stability of the luciferase reporter construct containing its 3’-UTR in a HIF-1 gene dependent manner. In contrast, two proteins regulating mRNA stability tristetrapolin (TTP), a member of the TISI CCCH zinc finger protein family, and PUM2, a homolog of Drosophila pumilio, were found to decrease the levels of endogenous DUSP6 mRNA and the activity of the DUSP6 in its 3’-UTR luciferase reporter construct in the HEK293 cell line68.
In the MAPK signaling pathway, DUSP6 undergoes highly specific interaction with only ERK1 and ERK2 at dual threonine and tyrosine residues of the TEY motif, which leads to DUSP6 inactivation69. Arkell et al.69reported that DUSP6 binds to ERK1/2 in both yeast and human cells, but fails to bind to ERK5. The same study demonstrated that recombinant ERK2 can induce catalytic activation of DUSP6,and DUSP6 expression can de-phosphorylate a co-expressed ERK2 construct and block the MEK1-driven activation of GAL4-ELK1, an ERK1/2-regulated transcription factor.However, because ERK5 is closely associated with ERK1/2 in the sense of catalytic domain and presence of the TEY motif for phosphorylation, and because it has similar function in cellular proliferation and survival, previous studies have suggested that ERK5 could be another substrate for DUSP670-75. This discordance demands further study.
A previously suggested spatiotemporal model can explain the mechanism of interaction between DUSP6 and ERK1/260,64. In this context, phosphorylation of DUSP6 and dephosphorylation of ERK1/2 would create a negative feedback loop to control ERK activity and indirectly the MAPK signal, which is vital for cell proliferation and differentiation (Figure 3)48. Two sites, serine 159 and 197, on DUSP6, have been identified as being involved in the phosphorylation that finally leads to degradation by proteasomes. The model clarifies the basis of the mutual interaction between DUSP6 and ERK1/2. In this respect, at the initial time point, active ERK1/2 is found mostly in the nucleus, rather than in the cytosol and ERK signal is maintained. Afterwards, the ERK signal begins to gradually diminish because of dephosphorylation of ERK1/2 by DUSP6. At a later point in time, however, active ERK begins to accumulate in the cytosol to re-sustain the signal. After signal recovery, active ERK begin to re-accumulate in the nucleus until the transmitted signal is diminished again and the same mechanism recurs60.
Interaction between DUSP6 and a well-known tumor suppressor gene, p53, occurs in a very unique manner and can determine cell’s fate. Genotoxic stress can spark cellular responses via p53 that result in cell cycle arrest and finally cell death. Piya et al.76reported that p53 inhibits the cell survival pathway by inducing DUSP6 upregulation, which enhances cell death through ERK 1/2 dephosphorylation, subsequently leading to Bcl-2 proteasomal degradation and Bad activation.This results in mitochondrial damage and eventually cell death76. These findings suggest that ERK protects cells from apoptosis by phosphorylating Bad, resulting in dissociation of Bad from Bcl-xL and protection of Bcl-2 from proteasomal degradation. Simultaneously, p53 also transactivates pro-death target genes, including Noxa, PUMA, PIG3, Bax, ASC,Fas, and DR5, converging their signals to enhance the cell death pathway76, as shown in Figure 4.
Another well-known target of ERK signaling is E twenty six(Ets) 2 protein, a transcription factor that can bind to the conserved region on the DUSP6 promoter in response to FGF or phorbol 12-myristate 13-acetate (PMA). Ets also can bind to intron 1 of DUSP6 in response to active ERK activity and to Ets-binding sites in the p53 promoter region, causing transactivation of p53 and leading to apoptosis; this interaction was observed in the thymus of Ets2 transgenic mice and in Ets2-overexpressing HeLa cells64,77-81. These findings establish a feedback regulatory loop among p53,DUSP6, Ets2, and ERK. In this scenario, Ets2 could be an activator for p53 and DUSP6, whereas p53 and DUSP6 may be inhibitors of Ets2 via ERK activation76.
Another gene linked to DUSP6 is BAG3, which encodes a multifunctional protein that contains the BAG domain,which can bind to heat shock protein (Hsp) 70 as well as to the other proteins via the WW domain with a proline-rich(PXXP) repeat and IPV (Ile-Pro-Val) motifs. Its functions include controlling levels, localization, and activity of other interacting proteins as well as regulating cellular pathways,such as apoptosis, autophagy, cytoskeleton organization and motility, and mechanotransduction83. Falco et al.84successfully identified a novel role of BAG3 in regulation of neo-angiogenesis and showed that its downregulation decreased angiogenesis. BAG3 was also found to interact with ERK and DUSP6, as removal of BAG3 resulted in reduced binding of DUSP6 to ERK and a subsequent sustained ERK level. This sustained ERK level caused reduction in phosphorylation of retinoblastoma (Rb) by cyclin D(Cdk4/5) and cyclin E (Cdk2), which in turn led to increased p21 and p15 levels and cell cycle arrest in the G1 phase. Thus,DUSP6 needs to form a complex with BAG3 to interact with phospho-ERK1/284.
DUSP6 has also been shown to interact with, and be phosphorylated by, protein kinase CK2α, a ubiquitous kinase. This was illustrated via the yeast two-hybrid system,which showed that DUSP6 can form a complex with CK2α,leading to modulation of ERK2-MAPK signaling and crossregulation of DUSP659. Serine-arginine protein kinase 1(SRPK1) is another gene that was found to interact with DUSP6. The signaling mechanism of SRPK1 in promoting gastric cancer progression was explored and it was suggested that DUSP6 might regulate the downstream effectors of SRPK185. Figure 4 shows a summary of the interaction between DUSP6 and associated genes.
Role of DUSP6 and its relevance in cancers
Previous research has reported that DUSP6 is associated with many types of cancer and that it plays a variety of roles. A tumor-suppressive role of DUSP6 via pivotal negativefeedback regulation of the ERK1/2 in the MAPK signaling pathway has been reported in many studies of diverse cancers, including pancreatic, lung, ovarian, esophageal squamous cell, and nasopharyngeal cancers40,86-88. However,the conventional hypothetical mechanism does not apply in all situations, and instead DUSP6 plays a tumor-promoting or pro-oncogenic role in cancers such as human glioblastoma, thyroid carcinoma, breast cancer, and acute myeloid leukemia8,89-92. In the case of melanoma, DUSP6 can have both functions depending on the histological subtypes of the cells93. Therefore, DUSP6 definitely influences and determines the fate of the carcinogenic pathway of a given cancer. The functions of DUSP6 are extensively explored below and are summarized in Table 2. Understanding the functions of DUPS6 and its modulation in cancer may lead to potential development of DUSP6 as a molecular target for various types of cancer.
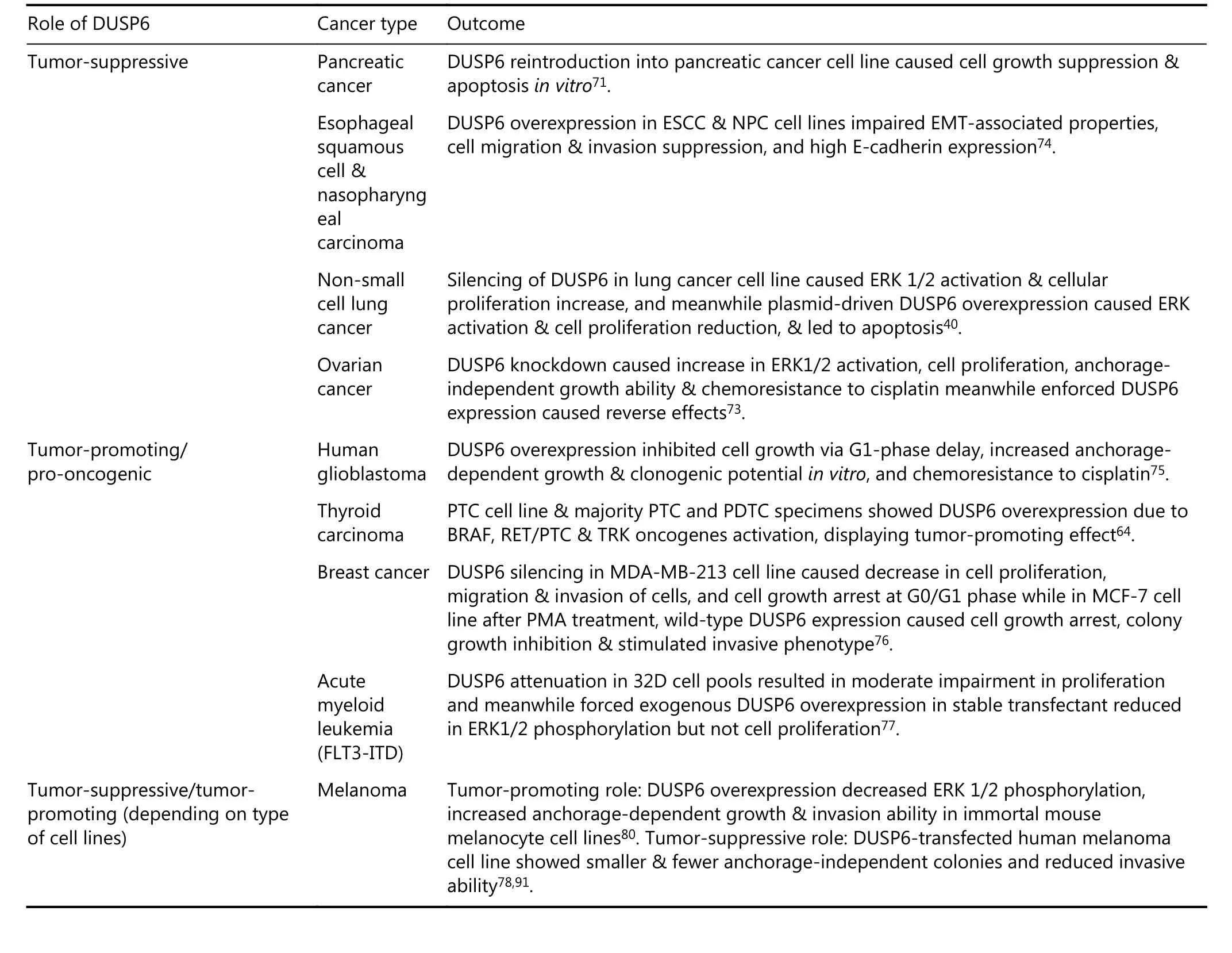
Table 2 Role of DUSP6 and its outcome in different types of cancers.
DUSP6 in pancreatic cancer
Furukawa and Horii95showed that there is loss of heterozygosity at 12q21 and 12q22.q23.1, where the DUSP6 gene resides. Molecular pathogenesis studies also revealed gain-of-function mutations of KRAS2, mostly in the pancreatic cell line. Mutation of KRAS2 coupled with DUSP6 loss caused uncontrolled cell growth94,95. An immunohistochemical investigation of a primary pancreatic cell line showed upregulation of the DUSP6 gene in mildly and severely dysplastic/in situ carcinoma but downregulation in invasive carcinoma, especially in the poorly differentiated type86. DUSP6 transcriptional suppression could be due to hypermethylation with histone deacetylation modification in the expressional control region in human pancreatic cancer96. Interestingly, reintroduction of DUSP6 via adenovirus and its exogenous expression caused cell growth suppression and apoptosis, thus displaying the tumorsuppressive role of the DUSP6 gene in pancreatic cancer in vitro86. The tumor-suppressive role of DUSP6 is also supported by other studies showing that restoration of chromosome 12 caused suppression of pancreatic cancer cell growth97and that exogenous overexpression of DUSP6 resulted in downregulation of MAPK1/ERK2 and AURKA as well as its related genes, such as AURKB, TPX2, and CENPA98.
DUSP6 in lung cancer
The tumor-suppressive role of DUSP6 was also observed in non-small cell lung cancers (NSCLCs)40,99-100. In microarray analysis of EGFR inhibition in EGFR-mutant NSCLC cells,several DUSPs, including DUSP6, were among the most highly and immediately regulated genes99. DUSP6 expression levels were weaker in most lung cancer cell lines than in human bronchial epithelial cells, and restoration of its expression suppressed cellular growth100. Screening of multiple NSCLC cell lines and primary human tumor specimens demonstrated that DUSP6 is expressed in tandem with ERK activation. The same study also showed dramatic downregulation of DUSP6 expression via pharmacologic inhibition of ERK activity40. Small-interfering RNA (siRNA)knockdown in a highly expressed DUSP6 lung cancer cell line showed a significant increase in ERK activation and cellular proliferation. In contrast, plasmid-driven overexpression in a stably transfected lowly expressed DUSP6 lung cancer cell line displayed significantly reduced ERK activation, cellular proliferation, and finally apoptosis40.
In addition to modulating oncogenesis, DUSP6 also influences the sensitivity of cancer cells to chemotherapy drugs either directly or indirectly. In lung adenocarcinoma cells, in which oncogenic rearrangement of the gene encoding for anaplastic lymphoma kinase (ALK), such as EML4-ALK,is observed, up to 40% of ALK+ patients failed to respond to ALK-inhibitor therapy101,102. Hrustanovic et al.103reported that downregulation of DUSP6 promoted resistance to ALK inhibitors in vitro as well as in individuals with EML4-ALK–positive lung adenocarcinoma by reactivation of the MAPK pathway. A similar study on CAR 2 and CAR 3 cells that have substantial basal phosphorylation of ERK revealed that stable reconstitution of DUSP6 restored sensitivity of the cells to crizotinib and the ability of crizotinib to suppress MAPK signaling. This finding highlights the potential use of DUSP6 as a MAPK kinase regulator in drug design or drug target strategies103.
DUSP6 in ovarian cancer
Loss of DUSP6 (MKP3) inflicted by ROS, which plays a tumor-suppressive role, was observed and linked to ovarian cancer progression. Previous research reported that more ROS are produced as ovarian cancer cells progress and develop, resulting in DUSP6 protein degradation via protein ubiquitination-proteosome; however, DUSP6 is restored after cells are treated with antioxidant87,104,105. The DUSP6 tumor-suppressive role in ovarian cancer was further shown by a knockdown study of endogenous DUSP6/MKP3 by short hairpin RNA (shRNA), which resulted in increased ERK1/2 activity, cell proliferation rate, anchorageindependent growth ability, and resistance to cisplatin in ovarian cancer cells. Conversely, enforced expression of DUSP6/MKP3 in DUSP6/MKP3-deficient ovarian cancer cells significantly reduced ERK1/2 activity and inhibited cell proliferation, anchorage-independent growth ability, and tumor development in nude mice. The enforced DUSP6/MKP3 expression also sensitized ovarian cancer cells to cisplatin-induced apoptosis in vitro and in vivo87.
DUSP6 in esophageal squamous cell carcinoma(ESCC) and nasopharyngeal carcinoma (NPC)
Significant loss of DUSP6 gene expression was found in 100% of 11 ESCC and 71% of 7 NPC cell lines. DUSP6 expression was also downregulated in 40% of 30 ESCC biopsies and 75% of 20 NPC biopsies in comparison to their respective normal counterparts. These results illustrate the tumor-suppressive role of DUSP6 in both cancers88. DUSP6 overexpression caused impairment of epithelialmesenchymal transition (EMT)-associated properties,polarized cell spheroid formation, increased expression of E-cadherin as EMT markers, and dramatic suppression of cell migration and invasion, further supporting the anti-tumor effects of DUSP688. DUSP6 expression was downregulated in ESCC cell lines and primary tumor specimens compared to their normal counterparts, and DUSP6 expression was also negatively associated with pathological grade. The same study also showed that exogenous DUSP6 expression significantly induced apoptosis, which illustrates the tumor-suppressive role of DUSP6 in ESCC carcinogenesis106.
DUSP6 in human glioblastoma
DUSP6 has been reported to have a tumor-promoting role in certain cases. Messina et al.89reported that upregulation of DUSP6 gene transcription in primary and long-term cultures of human glioblastoma plays a tumor-promoting role and exacerbates the malignant phenotype. DUSP6 overexpression in glioblastoma cultures through replication-deficient adenovirus-mediated expression of DUSP6 was found to inhibit cell growth by inducing G1-phase delay and increased anchorage dependent, as well as clonogenic potential in vitro.The study also showed that cisplatin treatment failed to sensitize DUSP6-overexpressing glioblastoma cells in vitro and in vivo, indicating chemoresistance, which normally causes poor treatment results in patients with glioblastoma.However, depletion by antisense DUSP6-AS reversed the effect by lowering the threshold sensitivity to drug treatment89. Another study revealed the relationship between glioblastoma multiforme and the EGFR gene. An EGFR mutant, which was likely to mediate high carcinogenic activity, caused upregulation of a small group of genes,including DUSP6, which influenced signaling pathways known to play a key role in oncogenesis and function in interconnected networks107.
DUSP6 in thyroid carcinoma
Papillary thyroid carcinoma (PTC) cell lines and the majority of PTC and poorly differentiated thyroid carcinoma (PDTC)specimens have demonstrated overexpression of DUSP6,which has as a tumor-promoting effect90. Activated BRAF,RET/PTC, and TRK oncogenes in thyroid carcinoma were able to upregulate DUSP6 expression and indirectly activate both the ERK1/2 pathway and its negative feedback mechanism. Levels of DUSP6 mRNA and protein were significantly higher and overexpressed in PTC and PDTC tissues, both in expression profile datasets and in patients’surgical samples, reflecting a positive correlation between DUSP6 expression and activation of the ERK1/2 pathway90.DUSP6 silencing resulted in reduction of branched morphogenesis in TPC1, reduced proliferation and enhanced apoptosis in the NIM-1 cell line, and reduced invasive ability of all four PTC cell lines, suggesting that DUSP6 plays a vital pro-tumorigenic role in thyroid carcinogenesis90.
DUSP6 in breast cancer
DUSP6 expression was reported to play an oncogenic role in breast cancers78,82,91. The specific role of the gene has been explored in triple-negative human breast cancer cells (MDAMB-231 cell), in which small interfering RNA (DUSP6-siRNA) and microRNA (miR-145) were employed to suppress the DUSP6 gene through transfection into the human breast cancer cells. This resulted in reduced DUSP6 mRNA and protein expression, cell proliferation, migration and invasion of the cells, and arrest at the G0/G1 phase91. In another study, DUSP6 was upregulated in the MCF-7 breast cancer cell line after being treated with PMA, which later inactivated ERK1/2 as a response to the sustained ERK 1/2 activation and played a positive role in proliferation and migration of the cells78. In contrast, silencing of DUSP6 accelerated cyclin-dependent kinase inhibitor upregulation(p21Waf1/Cip1), prevented cell growth arrest and inhibition of colony growth in soft agar, and stimulated the invasive phenotype. These findings illustrate the tumor-promoting role of DUSP678. DUSP6 was also found to be strongly upregulated in HER2-overexpressed breast cancer that promotes malignancy108. Another study reported that upregulation of DUSP6 rendered estrogen receptor-positive breast cancer cells resistant to the growth inhibitory effect of tamoxifen, illustrating the pro-oncogenic role of the phosphatase gene109.
DUSP6 in acute myeloid leukemia (AML)
The tumor-promoting role of DUSP6 has been demonstrated in AML92. The protein-tyrosine phosphatase (PTP)expression profile was established and the role of Fms-like tyrosine kinase 3 with internal tandem duplication (FLT3 ITD) was explored in AML cells from patients and cell lines that showed three transmembrane classical PTPs and six non-receptor classical PTPs, including several DUSPs, such as DUSP6. Pharmacological inhibition and silencing technology (si/shRNA) caused downregulation but increased constitutive ERK1/2 activation92. Attenuation of DUSP6 in 32D cell pools resulted in moderate impairment of proliferation, suggesting its positive role in FLT3 ITD-dependent cell growth. Forced exogenous DUSP6 overexpression in stable transfectants reduced ERK1/2 phosphorylation but did not inhibit cell proliferation,illustrating the DUSP6 pro-oncogenic role in FLT3 ITD-positive AML92.
DUSP6 in endometrial carcinoma
Endometrial carcinoma is a prevalent cancer in females, and epigenetic alterations have been found to play critical roles in the development and metastasis of endometrial cancer110,111.A DUSP6 gene is expressed in normal human uterine tissue112, and its expression is higher at stage I than stage II of endometrial cancer113. Recent intensive methylation profiling in 8 hyperplasia, 33 primary, and 53 metastatic endometrial cancers revealed the presence of hypomethylated CpG islands of the DUSP6 gene in primary and metastatic endometrial cancer, which supports the presence of epigenetic involvement in controlling the expression of DUSP6 in endometrial cancer113. Another study showed that DUSP6 is expressed at high levels in endometrial carcinoma, and an in vitro study using Ishikawa cells showed that overexpression of DUSP6 enhanced the growth-promoting effect of estrogen in the endometrial adenocarcinoma cells114. These results revealed the involvement of DUSP6 in the estrogen and estrogen receptor signaling pathway as well as the participation of DUSP6 as a negative feedback regulator of the MAPK/ERK1/2 signaling pathway in the development of endometrial adenocarcinoma114.
DUSP6 in malignant melanoma
The role of DUSP6 in malignant melanoma is very unique because it is histological subtype dependent93. Microarray data showed that 10% of the thick human primary melanoma had high DUSP6 expression and similar poor melanoma-specific survival, as the majority of thick primaries with low DUSP6 expression levels exhibited molecularly distinct melanoma subtype characteristics93. In another study, the tumor-suppressive role of DUSP6 in human malignant melanoma was observed when overexpression of DUSP6 reduced ERK activation, reduced basal or cisplatin-induced expression of the DNA repair gene ERCC1, and increased sensitivity of melanoma to cisplatin115.In tumorigenic mouse melanocyte cell lines of a mouse xenograft, a low level of ERK phosphorylation, high expression of DUSP6, low expression of p-MEK and p-AKT,and retention of p16 and PTEN were observed93. DUSP6 overexpression decreased ERK phosphorylation but increased anchorage-independent growth and invasive ability of their immortal mouse melanocytes. Ectopic expression of active MEK increased p-ERK levels, which suppress tumorigenic melanocyte invasiveness in a Transwell migration assay. These results illustrate the pro-oncogenic role of DUSP6 in mouse melanocyte cell lines93. In contrast,the same study also showed that a human malignant melanoma cell line that contains the BRAF V600E mutation and elevated ERK activity has smaller and fewer anchorageindependent colonies and decreased invasive ability in the DUSP6-transfected cells, illustrating the DUSP6 tumorsuppressive role.
Wittig-Blaich et al.116recently highlighted the oncogenic role of DUSP6 via ERK1/2-mediated phosphorylation. They analyzed four selected cancer cell lines, including melanoma,and constructed an isogenic single recombinant (ISR) of 108 genes in the melanoma cell line. The generated ISRs were subjected to cell cycle analyses, which demonstrated strong growth and a suppressive effect of five genes (DUSP6,RPS6KA2, MAPRE3, STARD3, and EMD). Further analysis showed consistent elevated levels of only DUSP6 mRNA expression in primary melanoma and melanoma cell lines compared to primary normal human epidermal melanocytes(NHEMs). The mRNA expression of the other four genes was not consistently elevated in primary melanoma and melanoma cell lines compared to NHEMs116. Analysis using siRNA-mediated knockdown of DUSP6 in BRAF wild-type melanoma resulted in either promotion of cell growth or no effect. However, knockdown of DUSP6 in BRAF V600E melanoma with high DUSP6 expression caused cell death via induction of apoptosis, which further illustrates the tumorsuppressive role of DUSP6116. BRAF V600E has been implicated in melanoma progression in several different mechanisms, such as by activation of the downstream MEK/ERK pathway, angiogenesis, evasion of senescence, and apoptosis117. In BRAF V600E melanoma, elevated expression of DUSP6 levels is required to compensate for BRAF V600E hyperactivation, which might otherwise trigger apoptosis via ERK1/2 downstream targets. This finding using BRAF V600E melanoma highlights the oncogenic role of DUSP6 via ERK1/2-mediated phosphorylation, and it successfully proves the concept that tumor suppressor DUSP6 could serve as a putative synthetic lethal target in melanoma with BRAF V600E mutations116.
Frontiers
This review illustrates that DUSP6 plays a very important role in carcinogenesis in numerous types of tumors and that it can act as both a tumor promoter and a tumor suppressor.Thus, this phosphatase gene has great potential as a prognostic factor, particularly in the evaluation of cancer treatment outcomes. DUSP6 also could be developed as a therapeutic target for future cancer treatment, either alone or coupled with current conventional treatments as an adjunct therapy. Another possible strategy for cancer treatment is to reverse or replace DUSP6 function. For example, Yokoyama et al.118demonstrated that increased ERK1/2 expression and downregulated DUSP6 caused pazopanib resistance in synovial sarcoma cells. Hence, simultaneous treatment with pazopanib and a MEK inhibitor would be a good strategy to overcome resistance. In another study, the DUSP6 gene was downregulated and became a vulnerable target of the skin tumor promoter palytoxin in initiated cells expressing oncogenic RAS119. In this case, DUSP6 may be a candidate target for upregulation in cells expressing oncogenic RAS.However, these approaches will require further detailed research.
Several characteristics of DUSP6 require further insight,such as how DUSP6 interacts with other family members of the phosphatase group in the sense of its physiological role and substrate specificity, and its interaction with other signaling pathways and metabolites. Another issue that needs further explanation is the reason for the different biochemical or biological roles of this enzyme in different cancers or in different histological subtypes of tissues of the same cancer93.
Conclusions
Current research on DUSP6 has provided clear clues about its role and mechanism in several cancers. This information will facilitate further advanced research on DUSP6 and its association in cancer as well as possible targeted cancer therapy.
Acknowledgements
This work was funded by a grant from Fundamental Research Grant Scheme, Ministry of Higher Education Malaysia(Grant No. 203.CIPPT.6711505).
Conflict of interest statement
No potential conflicts of interest are disclosed.
1.Dickinson RJ, Keyse SM. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci. 2006;119: 4607-15.
2.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004; 23: 2838-49.
3.Davis RJ. Signal transduction by the JNK group of MAP kinases.Cell. 2000; 103: 239-52.
4.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases.Science. 2002; 298: 1911-2.
5.Chang LF, Karin M. Mammalian MAP kinase signaling cascades.Nature. 2001; 410: 37-40.
6.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M,Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev.2001; 22: 153-83.
7.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update.Physiol Rev. 2012; 92: 689-737.
8.Dhillion AS, Hagan S, Rath O, Kolch W. MAP kinase signaling pathways in cancer. Oncogene. 2007; 26: 3279-90.
9.Lawrence MC, Jivan A, Shao CL, Duan LL, Goad D, Zaganjor E,et al. The roles of MAPKs in disease. Cell Res. 2008; 18: 436-42.
10.Sui XB, Kong N, Ye L, Han WD, Zhou JC, Zhang Q, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett.2014; 344: 174-9.
11.Treisman R. Regulation of transcription by MAP kinase cascades.Curr Opin Cell Biol. 1996; 8: 205-15.
12.Chen RE, Thorner J. Function and regulation in MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae.Biochim Biophys Acta. 2007; 1773: 1311-40.
13.Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71: 479-500.
14.Dhanasekaran DN, Johnson GL. MAPKs: function, regulation,role in cancer and therapeutic targeting. Oncogene. 2007; 26:3097-9.
15.Cowan KJ, Storey KB. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol. 2003; 206: 1107-15.
16.Zarubin T, Han JH. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2006; 15: 11-8.
17.Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signaling. Biochem J. 2010; 429: 403-17.
18.Fleming Y, Armstrong CG, Morrice N, Paterson A, Goedert M,Cohen P. Synergistic activation of stress-activated protein kinase 1/c-Jun N-terminal kinase (SAPK1/JNK) isoforms by mitogenactivated protein kinase kinase 4 (MKK4) and MKK7. Biochem J.2000; 352: 145-54.
19.Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta. 2007; 1773: 1341-8.
20.Cui J, Zhang M, Zhang YQ, Xu ZH. JNK pathway: diseases and therapeutic potential. Acta Pharmacol Sin. 2007; 28: 601-8.
21.Sabapathy K. Role of the JNK pathway in human diseases. Prog Mol Biol Trans Sci. 2012; 106: 145-69.
22.Huang D, Ding Y, Luo WM, Bender S, Qian CN, Kort E, et al.Inhibition of MAPK kinase signaling pathways suppressed renal cell carcinoma growth and angiogenesis in vivo. Cancer Res. 2008;68: 81-8.
23.Hu H, Han T, Zhuo M, Wu LL, Yuan CC, Wu LX, et al. Elevated COX-2 expression promotes angiogenesis through EGFR/p38-MAPK/Sp1-dependent signalling in pancreatic cancer. Sci Rep.2017; 4: 470
24.Ai XL, Chi Q, Qiu Y, Li HY, Li DJ, Wang JX, et al. Gap junction protein connexin43 deregulation contributes to bladder carcinogenesis via targeting MAPK pathway. Mol Cell Biochem.2017; 428: 109-18.
25.Dai Y, Yu C, Singh V, Tang L, Wang Z, McInistry R, et al.Pharmacological Inhibitors of the mitogen-activated protein kinase (MAPK) kinase/MAPK cascade interact synergistically with UCN-01 to induce mitochondrial dysfunction and apoptosis in human leukemia cells. Cancer Res. 2001; 61: 5106-15.
26.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002; 12: 9-18.
27.Robertson BW, Bonsal L, Chellaiah MA. Regulation of Erk1/2 activation by osteopontin in PC3 human prostate cancer cells.Mol Cancer. 2010; 9: 260
28.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EWT, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007; 1773: 1263-84.
29.Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors:potential targeting for therapeutic intervention. Leukemia. 2003;17: 1263-93.
30.Diehl NL, Enslen H, Fortner KA, Merritt C, Stetson N, Charland C, et al. Activation of the p38 mitogen-activated protein kinase pathway arrests cell cycle progression and differentiation of immature thymocytes in vivo. J Exp Med. 2000; 191: 321-34.
31.Lee K, Kenny AE, Rieder CL. P38 mitogen-activated protein kinase activity is required during mitosis for timely satisfaction of the mitotic checkpoint but not for the fidelity of chromosome segregation. Mol Biol Cell. 2000; 21: 2150-60.
32.Kitatani K, Wada M, Perry D, Usui T, Sun Y, Obeid LM, et al.Activation of p38 mitogen-activated protein kinase in gaucher’s disease. PLoS ONE. 2015; 10: e0136633
33.Ben-Hamo R, Efroni S. hsa-miR-9 and drug control over the P38 network as driving disease outcome in GBM patients. Syst Biomed. 2013; 1: 76-83.
34.Liu Z, Zheng Q, Chen WZ, Wu M, Pan GJ, Yang K, et al.Chemosensitizing effect of Paris Saponin I on Camptothecin and 10-hydroxycamptothecin in lung cancer cells via p38 MAPK, ERK,and Akt signaling pathways. Eur J Med Chem. 2017; 125: 760-9.
35.Chunga TW, Choib H, Lee JM, Ha SH, Kwak CH, Abekura F, et al. Oldenlandia diffusa suppresses metastatic potential through inhibiting matrix metalloproteinase-9 and intercellular adhesion molecule-1 expression via p38 and ERK1/2 MAPK pathways and induces apoptosis in human breast cancer MCF-7 cells. J Ethnopharmacol. 2017; 195: 309-17.
36.Waetzig GH, Seegert D, Rosenstiel P, Nikolaus S, Schreiber S. p38 mitogen-activated protein kinase is activated and linked to TNF-α signaling in inflammatory bowel disease. J Immunol. 2002; 168:5342-51.
37.Park SG, Kim SH, Kim KY, Yu SN, Choi HD, Kim YW, et al.Toyocamycin induces apoptosis via the crosstalk between reactive oxygen species and p38/ERK MAPKs signaling pathway in human prostate cancer PC-3 cells. Pharmacol Rep. 2017; 69: 90-6.
38.Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, et al. NF-κB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death.EMBO J. 2003; 22: 3898-909.
39.Lepage C, Léger DY, Bertrand J, Martin F, Beneytout JL, Liagre B.Diosgenin induces death receptor-5 through activation of p38 pathway and promotes TRAIL-induced apoptosis in colon cancer cells. Cancer Lett. 2011; 301: 193-202.
40.Zhang ZF, Kobayashi S, Borczuk AC, Leidner RS, LaFramboise T,Levine AD, et al. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis. 2010; 31: 577-86.
41.Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biol. 2002; 3: reviews3009.
42.Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14: 6-16.
43.Keyse SM. Protein phosphatases and the regulation of mitogenactivated protein kinase signalling. Curr Opin Cell Biol. 2000; 12:186-92.
44.Kidger AM, Keyse SM. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin Cell Dev Biol. 2016; 50: 125-32.
45.Huang CY, Tan TH. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012; 2: 24
46.Keyse SM, Ginsburg M. Amino acid sequence similarity between CL100, a dual-specificity MAP kinase phosphatase and cdc25.Trends Biochem Sci. 1993; 18: 377-8.
47.Bordo D, Bork P. The rhodanese/Cdc25 phosphatase superfamily:sequence-structure-function relations. EMBO Rep. 2002; 3: 741-6.
48.Kondoh K, Nishida E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim Biophys Acta. 2007; 1773: 1227-37.
49.Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene.2007; 26: 3203-13.
50.Karlsson M, Mathers J, Dickinson RJ, Mandl M, Keyse SM. Both nuclear-cytoplasmic shuttling of the dual specificity phosphatase MKP-3 and its ability to anchor MAP kinase in the cytoplasm are mediated by a conserved nuclear export signal. J Biol Chem. 2004;279: 41882-91.
51.Tárrega C, Ríos P, Cejudo-Marín R, Blanco-Aparicio C, van den Berk L, Schepens J, et al. ERK2 shows a restrictive and locally selective mechanism of recognition by its tyrosine phosphatase inactivators not shared by its activator MEK1. J Biol Chem. 2005;280: 37885-94.
52.Bermudez O, Pagès G, Gimond C. The dual-specificity MAP kinase phosphatases: critical roles in development and cancer. Am J Physiol Cell Physiol. 2010; 299: C189-202.
53.Farooq A, Chaturvedi G, Mujtaba S, Plotnikova O, Zeng L,Dhalluin C, et al. Solution structure of ERK2 binding domain of MAPK phosphatase MKP-3: structural insights into MKP-3 activation by ERK2. Mol Cell. 2001; 7: 387-99.
54.Denu JM, Dixon JE. Protein tyrosine phosphatases: mechanisms of catalysis and regulation. Curr Opin Chem Biol. 1998; 2: 633-41.
55.Stewart AE, Dowd S, Keyse SM, McDonald NQ. Crystal structure of the MAPK phosphatase Pyst1 catalytic domain and implications for regulated activation. Nat Struct Biol. 1999;6: 174-81.
56.Liu SJ, Sun JP, Zhou B, Zhang ZY, editors. Structural basis of docking interactions between ERK2 and MAP kinase phosphatase 3. Proc Natl Acad Sci USA. 2006; 103: 5326-31.
57.Tanoue T, Yamamoto T, Maeda R, Nishida E. A novel MAPK phosphatase MKP-7 acts preferentially on JNK/SAPK and p38α and β MAPKs. J Biol Chem. 2001; 276: 26629-39.
58.Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C, et al. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science. 1998; 280: 1262-5.
59.Castelli M, Camps M, Gillieron C, Leroy D, Arkinstall S, Rommel C, et al. MAP kinase phosphatase 3 (MKP3) interacts with and is phosphorylated by protein kinase CK2α. J Biol Chem. 2004; 279:44731-9.
60.Marchetti S, Gimond C, Chambard JC, Touboul T, Roux D,Pouysségur J, et al. Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol Cell Biol. 2005; 25: 854-64.
61.Bermudez O, Marchetti S, Pagès G, Gimond C. Post-translational regulation of the ERK phosphatase DUSP6/MKP3 by the mTOR pathway. Oncogene. 2008; 27: 3685-91.
62.Jurek A, Amagasaki K, Gembarska A, Heldin CH, Lennartsson J.Negative and positive regulation of MAPK phosphatase 3 controls platelet-derived growth factor-induced Erk activation. J Biol Chem. 2009; 284: 4626-34.
63.Ma L, Chen ZB, Erdjument-Bromage H, Tempst P, Pandolfi PP.Phosphorylation and functional inactivation of TSC2 by Erk:implications for tuberous sclerosis and cancer pathogenesis. Cell.2005; 121: 179-93.
64.Ekerot M, Stavridis MP, Delavaine L, Mitchell MP, Staples C,Owens DM, et al. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem J. 2008; 412: 287-98.
65.Eblaghie MC, Lunn JS, Dickinson RJ, Münsterberg AE, Sanz-Ezquerro JJ, Farrell ER, et al. Negative feedback regulation of FGF signaling levels by Pyst1/MKP3 in chick embryos. Curr Biol. 2003;13: 1009-18.
66.Kawakami Y, Rodríguez-León J, Koth CM, Büscher D, Itoh T,Raya Á, et al. MKP3 mediates the cellular response to FGF8 signalling in the vertebrate limb. Nat Cell Biol. 2003; 5: 513-9.
67.Gómez AR, López-Varea A, Molnar C, de la Calle-Mustienes E,Ruiz-Gómez M, Gómez-Skarmeta JL, et al. Conserved crossinteractions in Drosophila and Xenopus between Ras/MAPK signaling and the dual-specificity phosphatase MKP3. Dev Dyn.2005; 232: 695-708.
68.Bermudez O, Jouandin P, Rottier J, Bourcier C, Pagès G, Gimond C. Post-transcriptional regulation of the DUSP6/MKP-3 phosphatase by MEK/ERK signaling and hypoxia. J Cell Physiol.2011; 226: 276-84.
69.Arkell RS, Dickinson RJ, Squires M, Hayat S, Keyse SM, Cook SJ.DUSP6/MKP-3 inactivates ERK1/2 but fails to bind and inactivate ERK5. Cell Signal. 2008; 20: 836-43.
70.Nimesh M, Campbell DG, Morrice N, Peggie M, Cohen P. An analysis of the phosphorylation and activation of extracellularsignal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem J. 2003; 372:567-75.
71.Zhou GC, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995;270: 12665-9.
72.Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD.Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 1998; 395: 713-6.
73.Sarközi R, Miller B, Pollack V, Feifel E, Mayer G, Sorokin A, et al.ERK1/2-driven and MKP-mediated inhibition of EGF-induced ERK5 signaling in human proximal tubular cells. J Cell Physiol.2007; 211: 88-100.
74.Kamakura S, Moriguchi T, Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases: Identification and characterization of a signaling pathway to the nucleus. J Biol Chem. 1999; 274: 26563-71.
75.Zou GM, Chen JJ, Ni J. LIGHT induces differentiation of mouse embryonic stem cells associated with activation of ERK5.Oncogene. 2006; 25: 463-9.
76.Piya S, Kim JY, Bae J, Seol DW, Moon AR, Kim TH. DUSP6 is a novel transcriptional target of p53 and regulates p53-mediated apoptosis by modulating expression levels of Bcl-2 family proteins. FEBS Lett. 2012; 586: 4233-40.
77.Znosko WA, Yu SB, Thomas K, Molina GA, Li CJ, Tsang W, et al.Overlapping functions of Pea3 ETS transcription factors in FGF signaling during zebrafish development. Dev Biol. 2010;342: 11-25.
78.Nunes-Xavier CE, Tárrega C, Cejudo-Marín R, Frijhoff J, Sandin Å, Östman A, et al. Differential up-regulation of MAP kinase phosphatases MKP3/DUSP6 and DUSP5 by Ets2 and c-Jun converge in the control of the growth arrest versus proliferation response of MCF-7 breast cancer cells to phorbol ester. J Biol Chem. 2010; 285: 26417-30.
79.Furukawa T, Tanji E, Xu SH, Horii A. Feedback regulation of DUSP6 transcription responding to MAPK1 via ETS2 in human cells. Biochem. Biophys Res. Commun. 2008; 377: 317-20.
80.Venanzoni MC, Robinson LR, Hodge DR, Kola I, Seth A. ETS1 and ETS2 in p53 regulation: spatial separation of ETS binding sites(EBS) modulate protein: DNA interaction. Oncogene. 1996; 12:1199-204.
81.Wolvetang EJ, Wilson TJ, Sanij E, Busciglio J, Hatzistavrou T, Seth A, et al. ETS2 overexpression in transgenic models and in Down syndrome predisposes to apoptosis via the p53 pathway. Hum Mol Genet. 2003; 12: 247-55.
82.Hagan CR, Knutson TP, Lange CA. A Common Docking Domain in Progesterone Receptor-B links DUSP6 and CK2 signaling to proliferative transcriptional programs in breast cancer cells.Nucleic Acids Res. 2013; 41: 8926-42.
83.De Marco M, Basile A, Iorio V, Festa M, Falco A, Ranieri B, et al.Role of BAG3 in cancer progression: A therapeutic opportunity.Semin Cell Dev Biol. 2017; in press.
84.Falco A, Festa M, Basile A, Rosati A, Pascale M, Florenzano F,et al. BAG3 controls angiogenesis through regulation of ERK phosphorylation. Oncogene. 2012; 31: 5153-61.
85.Xu XT, Wei YH, Wang SD, Luo M, Zeng H. Serine-arginine protein kinase 1 (SRPK1) is elevated in gastric cancer and plays oncogenic functions. Oncotarget. 2017; 8: 61944-57.
86.Furukawa T, Sunamura M, Motoi F, Matsuno S, Horii A.Potential tumor suppressive pathway involving DUSP6/MKP-3 in pancreatic cancer. Am J Pathol. 2003; 162: 1807-15.
87.Chan DW, Liu VWS, Tsao GSW, Yao KM, Furukawa T, Chan KKL, et al. Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells.Carcinogenesis. 2008; 29: 1742-50.
88.Wong VCL, Chen H, Ko JMY, Chan KW, Chan YP, Law S, et al.Tumor suppressor dual-specificity phosphatase 6 (DUSP6)impairs cell invasion and epithelial-mesenchymal transition(EMT)-associated phenotype. Int J Cancer. 2012; 130: 83-95.
89.Messina S, Frati L, Leonetti C, Zuchegna C, Di Zazzo E, Calogero A, et al. Dual-specificity phosphatase DUSP6 has tumorpromoting properties in human glioblastomas. Oncogene. 2011;30: 3813-20.
90.Degl'Innocenti D, Romeo P, Tarantino E, Sensi M, Cassinelli G,Catalano V, et al. DUSP6/MKP3 is overexpressed in papillary and poorly differentiated thyroid carcinoma and contributes to neoplastic properties of thyroid cancer cells. Endocr Relat Cancer.2013; 20: 23-37.
91.Song HM, Wu CY, Wei CK, Li DF, Hua KY, Song JL, et al.Silencing of DUSP6 gene by RNAi-mediation inhibits proliferation and growth in MDA-MB-231 breast cancer cells: an in vitro study. Int J Clin Exp Med. 2015; 8: 10481-90.
92.Arora D, Köthe S, van den Eijnden M, van Huijsduijnen RH,Heidel F, Fischer T, et al. Expression of protein-tyrosine phosphatases in Acute Myeloid Leukemia cells: FLT3 ITD sustains high levels of DUSP6 expression. Cell Commun Signal. 2012;10: 19
93.Li WL, Song L, Ritchie AM, Melton DW. Increased levels of DUSP6 phosphatase stimulate tumourigenesis in a molecularly distinct melanoma subtype. Pigment Cell Melanoma Res. 2012;25: 188-99.
94.Caldas C, Kern SE. K-ras mutation and pancreatic adenocarcinoma. Int J Pancreatol. 1995; 18: 1-6.
95.Furukawa T, Horii A. Molecular pathology of pancreatic cancer:in quest of tumor suppressor genes. Pancreas. 2004; 28: 253-6.
96.Xu SH, Furukawa T, Kanai N, Sunamura M, Horii A. Abrogation of DUSP6 by hypermethylation in human pancreatic cancer. J Hum Genet. 2005; 50: 159-67.
97.Furukawa T, Sunamura M, Horii A. Molecular mechanisms of pancreatic carcinogenesis. Cancer Sci. 2006; 97: 1-7.
98.Furukawa T, Kanai N, Shiwaku HO, Soga N, Uehara A, Horii A.AURKA is one of the downstream targets of MAPK1/ERK2 in pancreatic cancer. Oncogene. 2006; 25: 4831-9.
99.Kobayashi S, Shimamura T, Monti S, Steidl U, Hetherington CJ,Lowell AM, et al. Transcriptional profiling identifies cyclin D1 as a critical downstream effector of mutant epidermal growth factor receptor signaling. Cancer Res. 2006; 66: 11389-98.
100.Okudela K, Yazawa T, Woo T, Sakaeda M, Ishii J, Mitsui H, et al.Down-regulation of DUSP6 expression in lung cancer: its mechanism and potential role in carcinogenesis. Am J Pathol.2009; 175: 867-81.
101.Shaw AT, Kim DW, Mehra R, Tan DSW, Felip E, Chow LQM, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014; 370: 1189-97.
102.Camidge DR, Doebele RC. Treating ALK-positive lung cancer—early successes and future challenges. Nat Rev Clin Oncol. 2012; 9: 268-77.
103.Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S,Blakely CM, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK–positive lung cancer. Nat Med. 2015; 21: 1038-47.
104.Inukai N, Yamaguchi Y, Kuraoka I, Yamada T, Kamijo S, Kato J,et al. A novel hydrogen peroxide-induced phosphorylation and ubiquitination pathway leading to RNA polymerase II proteolysis.J Biol Chem. 2004; 279: 8190-5.
105.Cao C, Li YP, Leng YM, Li P, Ma QJ, Kufe D. Ubiquitination and degradation of the Arg tyrosine kinase is regulated by oxidative stress. Oncogene. 2005; 24: 2433-40.
106.Ma J, Yu X, Guo L, Lu SH. DUSP6, a tumor suppressor, is involved in differentiation and apoptosis in esophageal squamous cell carcinoma. Oncol Lett. 2013; 6: 1624-30.
107.Waha A, Felsberg J, Hartmann W, von dem Knesebeck A, Mikeska T, Joos S, et al. Epigenetic downregulation of mitogen-activated protein kinase phosphatase MKP-2 relieves its growth suppressive activity in glioma cells. Cancer Res. 2010; 70: 1689-99.
108.Lucci MA, Orlandi R, Triulzi T, Tagliabue E, Balsari A, Villa-Moruzzi E. Expression profile of tyrosine phosphatases in HER2 breast cancer cells and tumors. Cell Oncol. 2010; 32: 361-72.
109.Cui YK, Parra I, Zhang M, Hilsenbeck SG, Tsimelzon A,Furukawa T, et al. Elevated expression of mitogen-activated protein kinase phosphatase 3 in breast tumors: a mechanism of tamoxifen resistance. Cancer Res. 2006; 66: 5950-9.
110.Caplakova V, Babusikova E, Blahovcova E, Balharek T, Zelieskova M, Hatok J. DNA methylation machinery in the endometrium and endometrial cancer. Anticancer Res. 2016; 36: 4407-20.
111.Banno K, Yanokura M, Iida M, Masuda K, Aoki D. Carcinogenic mechanisms of endometrial cancer: involvement of genetics and epigenetics. J Obstet Gynaecol Res. 2014; 40: 1957-67.
112.Expressed Sequence Tag Database.http://www.ncbi.nlm.nih.gov/UniGene/ESTProfileViewer.cgi?ugli st=Hs.298654
113.Wu HJ, Chen YP, Liang J, Shi B, Wu G, Zhang Y, et al.Hypomethylation-linked activation of PAX2 mediates tamoxifenstimulated endometrial carcinogenesis. Nature. 2005; 438: 981-7.
114.Zhang H, Guo QF, Wang C, Yan L, Fu YB, Fan MJ, et al. Dualspecificity phosphatase 6 (Dusp6), a negative regulator of FGF2/ERK1/2 signaling, enhances 17β-estrodial-induced cell growth in endometrial adenocarcinoma cell. Mol Cell Endocrinol.2013; 376: 60-9.
115.Li W, Melton DW. Cisplatin regulates the MAPK kinase pathway to induce increased expression of DNA repair gene ERCC1 and increase melanoma chemoresistance. Oncogene. 2012; 31:2412-22.
116.Wittig-Blaich S, Wittig R, Schmidt S, Lyer S, Bewerunge-Hudler M, Gronert-Sum S, et al. Systematic screening of isogenic cancer cells identifies DUSP6 as context-specific synthetic lethal target in melanoma. Oncotarget. 2017; 8: 23760-74.
117.Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene. 2011; 30: 3477-88.
118.Yokoyama N, Matsunobu T, Matsumoto Y, Fukushi JI, Endo M,Hatano M, et al. Activation of ERK1/2 causes pazopanib resistance via downregulation of DUSP6 in synovial sarcoma cells. Sci Rep.2017; 7: 45332
119.Warmka JK, Mauro LJ, Wattenberg EV. Mitogen-activated protein kinase phosphatase-3 is a tumor promoter target in initiated cells that express oncogenic Ras. J Biol Chem. 2004; 279:33085-92.
杂志排行

Cancer Biology & Medicine的其它文章
- The evolution of Epstein-Barr virus detection in nasopharyngeal carcinoma
- Silencing of syndecan-binding protein enhances the inhibitory effect of tamoxifen and increases cellular sensitivity to estrogen
- EGFR tyrosine kinase inhibitor HS-10182 increases radiation sensitivity in non-small cell lung cancers with EGFR T790M mutation
- Calcium channel α2δ1 subunit as a novel biomarker for diagnosis of hepatocellular carcinoma
- Parkin protein expression and its impact on survival of patients with advanced colorectal cancer
- A new combined criterion to better predict malignant lesions in patients with pancreatic cystic neoplasms