高通量转录组测序技术在猪肉品质研究中的应用进展
2018-03-07尹梦迪黄永震王周利姜艳芬郭抗抗王晶钰张彦明
尹梦迪,黄永震,王周利,姜艳芬,郭抗抗,王晶钰,张彦明,贺 花
(1.西北农林科技大学动物医学院,陕西杨凌 712100;2.西北农林科技大学食品科学与工程学院,陕西杨凌712100;3.西北农林科技大学动物科技学院,陕西杨凌 712100)
中国是世界上最大的猪肉生产和消费国,中国猪肉消费量约占全球猪肉消费量的一半。 随着我国国民对猪肉类需求量的提高,猪的日增重、饲料转化率及抗病性等显著提高,然而肉品质却显著下降。随着消费者对猪肉品质的日益注重,仅在饲养中改变猪的进食营养配比已不能从根本上解决问题,只有对种猪进行改良才能从根本上解决肉的品质问题,因此种猪改良迫在眉睫。现有的传统人工选育方法表现出时间跨度长、强度高等缺陷,因而在科学研究上,猪的遗传改良有很大的发展空间。
猪肉品质常用嫩度、持水力、风味、香味和营养成分等进行评定。大多数决定肉质性状的肌纤维(MF)特性具有遗传效应,受基因调控[1]。目前,已确定影响肉品质的主效基因仅为氟烷基因和RN-基因[2]。为了发现更多的影响肉品质的主效基因,可以应用高通量测序(High-Throughput Sequencing)技术以对一个物种的基因组与转录组进行深入全貌的分析,从而帮助科学家找到更多影响猪肉品质的基因。作为基因组研究的重要补充,蛋白质测序(Protein Sequencing)是另一重要研究领域。近年来,旨在阐明生物体全部基因组、转录组、蛋白质组的研究工作开始大规模开展。将高通量的测序技术应用于猪肉品质分析,利用各组学技术寻找、筛选并鉴定与肉品质有关的标记基因与蛋白,整合基因组数据和蛋白质组数据,可帮助研究人员研究影响猪肉品质的遗传水平因素,进而扩展应用于畜类育种工作中。本文综述了高通量转录组测序技术在研究猪肉品质相关的肌内脂肪(IMF)和MF转录组的应用,重点阐述了其研究进展。
1 高通量测序技术及其比较
高通量测序具有同时对几十万到几百万条DNA或RNA分子进行并行测序的功能。第二代测序技术广泛应用于寻找候选基因,是目前组学研究中的主要技术。高通量测序分类:在基因组水平上,对没有参考序列的物种进行从头测序(Denovo Sequencing),对有参考序列的物种,进行全基因组重测序(Resequencing);在转录组水平上,进行全转录组测序(Whole Transcriptome Resequencing),或者进行小分子RNA测序(Small RNA Sequencing)。为了对基因组上的甲基化位点和与特定转录因子结合的DNA区域进行检测,可以采用转录组测序与染色质免疫共沉淀(ChIP)技术同时结合甲基化DNA免疫共沉淀(MeDIP)技术。
Roche公司的454焦磷酸测序技术、ABI公司的SOLiD技术和Illumina公司的Solexa技术是具有代表性的第二代测序技术。454焦磷酸测序读长最长,当读长超过400 bp时,454焦磷酸测序的准确性仍在99%以上,因而应用最广泛,但是有误差随同聚物增长而增大的局限,且通量低,费用高,使得该测序平台的市场化普及被限制,常应用在对未知基因组进行从头测序、获得新基因的宏基因组学(Metagenomics)及微生物多样性等领域[3];Solexa技术序列读长相对454测序较短,费用也低,同时精确度高,样品需求量低,操作简便快速,缺点是判断AT富集区域的能力低,所以应用面相较于其他技术较窄,适合基因组重测序、RNA测序、表观遗传学研究[4];SOLiD技术测序读数最短,但扩展性较强,准确率高,由于存在着参考序列和读长无法匹配的可能,常应用于小RNA和全转录组等高敏感检测,同时由于其双碱基编码和校正系统的原理与重测序相似,应用于具有高质量参考基因组序列物种的重测序和SNP检测等[5](表1)。
尽管新一代测序平台已经显著降低了测序费用,然而完整的基因组测序费用仍然很高。除了费用高,研究人员还发现很难从大量变异中鉴定出真正起作用的位点。因此产生了最初就关注编码区变异的外显子捕获技术,其利用外显子捕获芯片从基因组DNA中抓出外显子部分后测序,但费用仍不低。转录组测序(Transcriptome Sequencing)又称RNA-seq,继而成为基因组测序的替代,与基因组测序相比,转录组测序的弊端是会遗漏一些低表达基因,优点是获得了更多的如基因表达水平和剪接模式的信息。因此在有高表达基因的组织来源并执行适当的质量控制筛选前提下,转录组测序是一种快速廉价的替代方法[6]。高通量转录组测序在研究真核生物的基因表达调控和遗传育种等方面具有不可估量的潜力。
转录组是指在某一特定发育时期或者某一生理条件下,细胞内所有转录产物的集合,包括mRNA和非编码RNA。转录组研究是基因功能及结构研究的基础和出发点。转录组测序的研究对象为特定细胞在某一功能状态下所能转录出来的所有RNA的总和。从总RNA中富集出单链mRNA,再反转录成双链cDNA,随后进行高通量测序,并与基因组DNA序列进行比对。对于基因组序列信息已知的生物,依据现有的基因注释结果,分析基因在不同组织中的差异表达情况、选择性剪切(Alternative Splicing,AS)、单核苷酸多态性(Single Nucleotide Polymorphisms,SNPs)和基因融合等特征。主要步骤:①建库。提取样品总RNA,利用脱氧核糖核酸酶(DNAaseI)除去样品中滞留的DNA,后用含多聚体的磁珠对mRNA分离纯化。纯化后的RNA序列被随机打断成小片段,反转录合成cDNA。②上机测序。一般采用双向测序以提高准确度[7]。③生物信息学分析。将数以百万计的reads与参考基因组进行序列比对,进行基因的表达注释、新转录本预测、SNP[8]以及AS[9]等分析。
在众多的转录组学分析方法中,转录组测序技术以新一代高通量测序技术为基础,是一个高度灵活的平台,与其他转录组学技术比较,具有通量高、速度快、成本低、灵敏度高的优势,可以获得低丰度的表达基因,不局限于已知的基因组序列信息,不需要克隆的步骤,操作简单,应用领域广。近年来,随着转录组学与高通量测序技术的高速发展,目前已超越传统的基于杂交技术的芯片法和基于一代 Sanger测序的SAGE、MPSS、全长cDNA文库、EST文库等方法,成为转录组分析的主要手段。
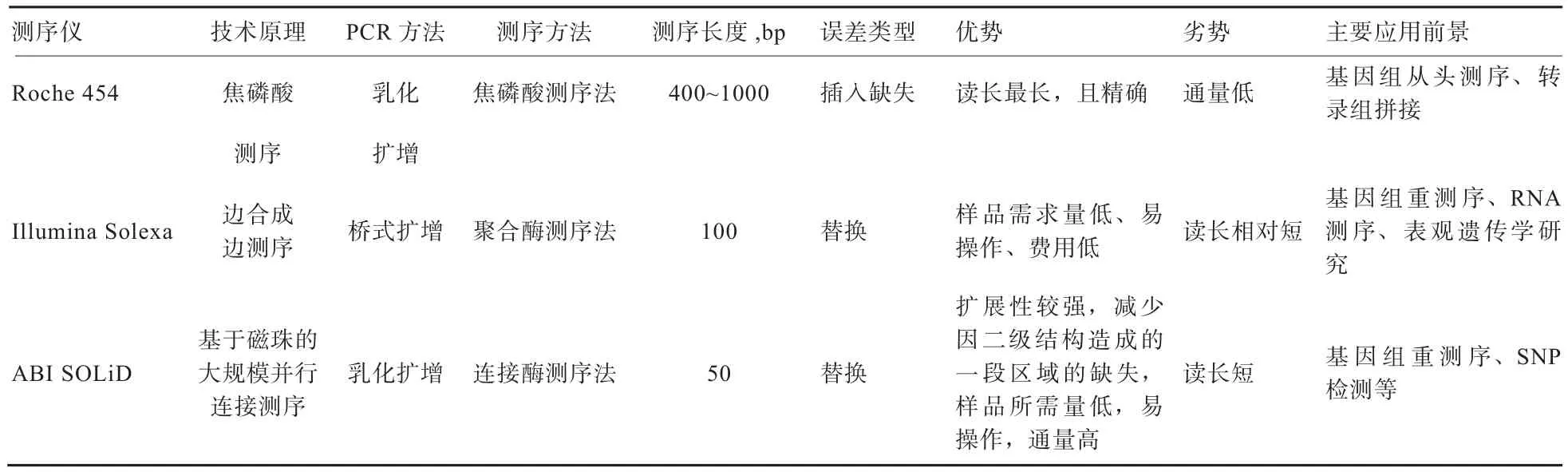
表1 不同测序平台的高通量测序技术特点比较[2-5]
2 高通量测序技术在IMF中的研究进展
IMF存在于猪的MF内部和肌束之间,在相当大的程度上决定着肉品质。适度的脂肪含量可使肌肉口感细腻、香味浓郁。2%~3%的IMF含量对猪肉的食用特性比较理想。IMF与系水力、风味和嫩度有很强的相关性。随着肌内脂肪含量的增多,结缔组织的物理强度逐渐降低,肉的嫩度得到改善[10]。谭林等[11]发现,在适当的范围内(1%~3%),随着IMF含量的升高,猪肉的大理石纹评分增加,肉的系水力得到改善(表2)。

表2 猪的IMF含量与大理石纹评分[11]
中外猪种在产肉性状上差异较大,且存在互补性。美国汉普夏猪、杜洛克猪、巴克夏猪、英国大白猪、比利时皮特兰猪和丹麦长白猪等具有瘦肉率高且生长较快的优势,但肉质风味较差;我国地方猪种一般生长慢、瘦肉率低,但肉质相对较好。荣昌猪的IMF含量、肉色和大理石纹评分显著高于杜洛克猪,总氨基酸含量和呈味氨基酸总量显著高于杜洛克猪,背最长肌的饱和脂肪酸含量显著低于杜洛克猪,单不饱和脂肪酸含量显著高于杜洛克猪[12]。肌肉和脂肪细胞均来源于中胚层的共同祖先细胞,其发育是动态调控过程,可互相转换,但关键基因的相互作用机制目前仍然不清楚。
数字化基因表达谱(Digital Gene Expression Profiling,DGEP)结合了新一代高通量测序技术和高性能计算分析技术,可以用来捕捉一个特定组织的整个转录组[13]。细胞数目的增多是导致IMF组织发育的决定性因素。肌肉组织中存在肌卫星细胞(MSCs)和脂肪来源干细胞(ADSCs)2种干细胞群体,体外的研究表明这2种干细胞群体均可以分化为脂肪细胞[14]。张凤[15]利用肌肉生长抑制素(Myostatin,MSTN)处理ADSCs和MSCs,利用Illumina Solexa测序技术对猪的2种干细胞中的RNA进行有参考基因组的表达谱测序,通过比较2种细胞处理前后的差异表达基因,获得与脂肪发育相关的基因WISP2和KLF6,并通过构建超表达载体和合成干涉片段,分别从功能获得和功能缺失两方面进一步研究,确定KLF6基因促进细胞的成脂分化,而WISP2基因抑制细胞的成脂分化,该结果为研究KLF6的功能提供了基础数据,也为揭示猪脂质代谢的调控机制提供了试验参考。KLF6是KLFs家族的成员,可通过与靶基因启动子上的GC框或CACCC元件相结合从而调控下游众多靶基因的表达,前期许多研究发现KLF6基因与脂肪肝的产生有密切关系[16]。近些年有研究发现,KLF6参与到机体前体脂肪细胞的分化且对脂肪细胞成脂分化起正向调控作用[15,17]。KLF6基因在畜禽类中与牛具有最近的亲缘关系,其次为山羊、绵羊、猪,与鸡具有最远的亲缘关系[18]。由此可以推断,KLF6基因不仅在猪中,还有极大可能在大部分畜类脂质代谢的调控中起作用。
邢凯等[19]利用转录组测序研究了长白猪、松辽黑猪的脂肪和肝脏组织,筛选出背膘厚极端表型个体间的多个调节脂肪代谢的差异基因,初步找到多个调控脂肪沉积的重要基因和通路,证明了肝脏在动物体内脂肪代谢调控中的关键作用。Li等[20]利用miRNA-seq从7、240日龄的荣昌猪背部皮下脂肪中检测出miR-143、miR-103、miR-148和Let-7等miRNA,发现仔猪脂肪组织中miR-378表达量最高,成年猪脂肪组织中miR-143表达量最高,且仔猪脂肪组织中miR-143、miR-378、miR-103、miR-148a、miR-21、miR-10b、miR-30a-5p和miR-199a-3p均高表达于2个发育阶段的背部皮下脂肪组织。
比较中外品种间不同发育点的差异是寻找关键基因的一个重要途径。研究人员可以利用高通量测序技术绘制中外猪种生长发育时期肌肉的转录组和猪的发育性图谱,分析标签和转录本的表达模式,寻找重要候选基因,鉴定与肉品质相关重要基因在中外猪种之间存在的差异。张冬杰等[21]构建了民猪(地方脂肪型)和大白猪(西方瘦肉型)背最长肌组织的数字化基因表达谱(Digital Gene Expression Profiling,DGEP),研究了11个可能与肉品质相关的显著差异基因,对比发现差异基因中FAS、FABP4和Cav-1在脂肪型民猪中的表达水平显著高于瘦肉型大白猪表达量,推断FAS基因和FABP4基因为与IMF含量正相关的基因。高通量测序在猪IMF相关基因挖掘中的研究进展见表3。

表3 高通量测序在猪的IMF相关基因挖掘中的研究进展
3 高通量测序技术在MF中的研究进展
MF是肌肉的基本构成单位,占畜类肌肉组织的75%~90%,MF的形态特征是肌肉特性的主要决定因素。其中,MF总数、横截面积和长度是MF的3个重要特征指标。不同品种间MF含量不同,MF含量是影响猪肉品质的重要因素,并可以稳定遗传[24]。骨骼肌中纤维类型与肉品质之间存在密切的关系,而纤维的多样性是由于肌肉组织中蛋白(如肌球蛋白重链、肌动蛋白、肌联蛋白)的不同表达导致,600~800 ku的伴肌动蛋白和超过1 200 ku的肌联蛋白都会在骨骼肌中特异性大量表达[25]。Larzul等[26]研究发现,遗传因素是影响MF类型的主要非营养因素,不同品种猪的MF组成有差异且具有较高的遗传性,其中1型纤维和2b型纤维的比例都具有高度的遗传力(h=0.46,h=0.58),通过遗传育种可以选择性地降低MF的收缩性能,选择性地降低肌肉横断面中肌红纤维的比例,最终通过降低宰后pH下降的速率和程度来改善肉质。Kim等[27]基于主成分分析发现,猪品种之间有明显的特性差异,眉山猪的肉品质明显劣于其他品种,而杂交品种和巴克夏猪肌纤维的性能更优,表现出更好的肉品质。不同品种猪的MF与肉品质主成分分析见表4。
由上述可知,MF的类型、粗细、长短直接影响肉的口感。彭兴[28]构建了不同发育时间点的猪骨骼肌小RNA文库,结合Solexa高通量测序结果,初步认定TSC1和SMAD7基因均为miRNA-21的靶标且为miRNA-21下调,miRNA-21可能通过靶向结合TSCl以及SMAD7参与调控mTOR信号通路以及TGF-β信号通路来影响肌肉增殖分化,参与骨骼肌的生长调控。Hou等[29]采用Solexa测序技术在通城猪背最长肌混合RNA文库中检测出275个miRNAs,其中miR-1、miR-378和miR-206表达量最高,且miR-378在胚胎期第33天低表达、第65~90天逐渐增加,出生时达最高水平,随后相对稳定,进一步研究发现miR-378与骨形成蛋白2(BMP)和丝裂原活化蛋白激酶1(MAPK1)的增殖和分化均有关。朱嘉宇[30]构建了猪不同类型肌肉组织的基因表达文库,采用转录组测序技术检测基因表达谱,对其功能进行聚类,Pathway分析,调控网络构建,并进行了新转录本预测、基因结构优化、可变剪切分析以及SNP分析,发现SUFU基因的3´UTR区域存在miR-423-5p的结合位点,干扰SUFU能够抑制C2C12成肌细胞的增殖和分化;miR-423-5p的表达量随着C2C12细胞分化逐渐上升,在分化的后期达到顶峰,然后逐渐下降;超表达miR-423-5p能够抑制C2C12细胞分化,并且抑制肌管形成。该研究结果为肌肉发育和MF类型的调控机制提供了更加深入的认识,为进一步研究肉品质相关的基因提供了很好的参考。

表4 不同品种猪的主成分分析[27]
根据表5中相关研究可知,控制细胞的分化方向的基因及通路一旦找到,即可通过操控该基因来控制分化的方向从而在源头上改变IMF及MF的特征。脂肪沉积通路从脂肪沉积的方向影响IMF。前期大量研究发现,控制激素及日粮水平可调节畜禽基因表达。后期研究应致力于探索通过控制激素水平及日粮中的营养成分来控制含FAS基因及KLF6基因在内的IMF相关基因、TSC1、SMAD7及miR-423-5p在内的肌肉发育相关基因转录,控制RNA稳定性及翻译水平,有效地控制脂肪与MF在体内的合成及代谢。随着对IMF与肌肉相关基因及通路的不断挖掘,当脂肪与肌肉含量达到可控,即可向市场上提供规范与细化的肉类品种,满足消费者多样的需求。
4 研究展望
目前转录组测序技术仍然面临挑战。基于长读长的3代测序技术虽然发展迅速,可测序错误率仍然偏高。目前比较好的做法是将3代测序和2代测序相结合,提高转录组测序的准确性。目前单细胞RNA-seq(Single-Cell RNA-seq)技术为小样本量的转录组分析提供了有效的分析手段。特别是对于异质性较强的组织样本,需要在单细胞水平进行转录组分析,单细胞转录组测序显示了其独特的优势。
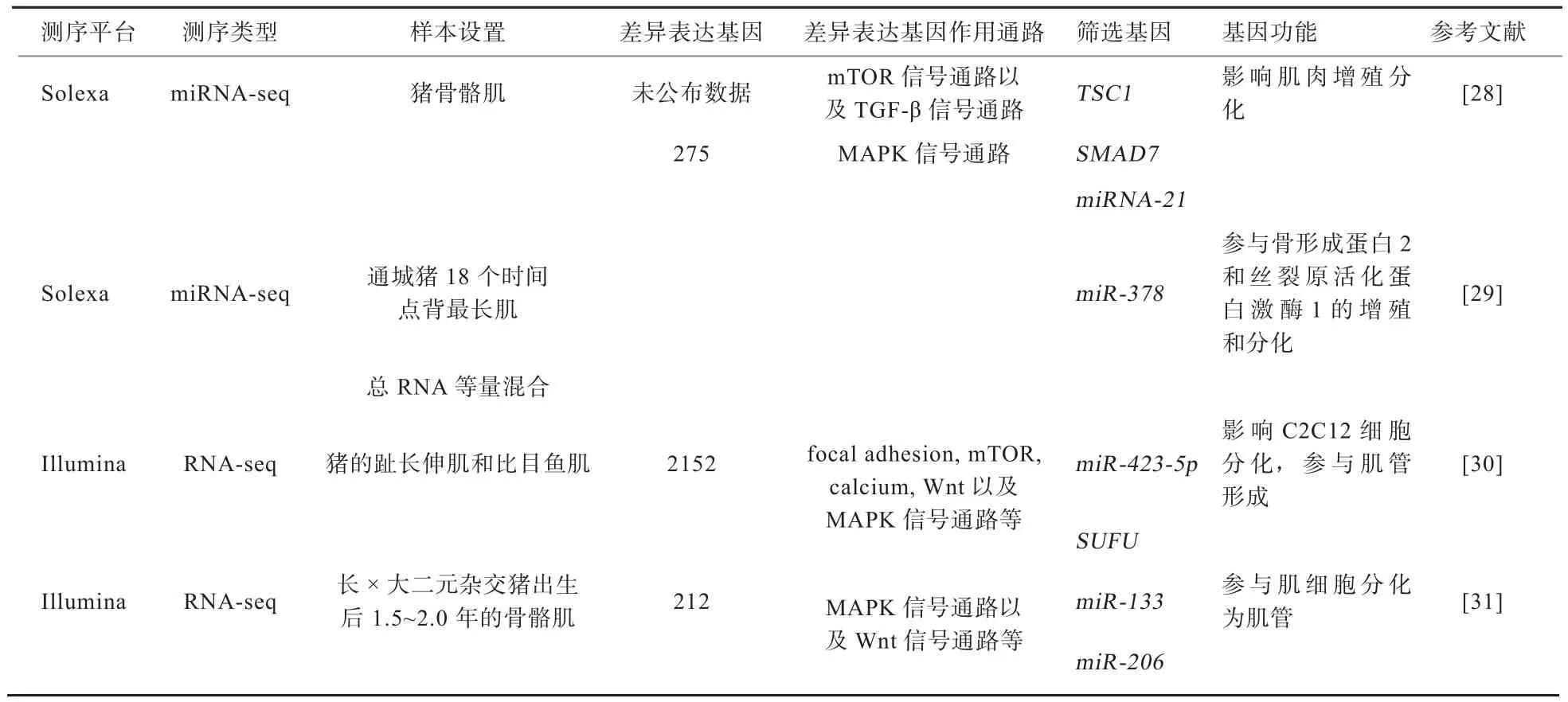
表5 高通量测序在猪的MF相关基因挖掘中的研究进展
伴随高通量测序技术的快速发展,测序费用急剧下降。虽然高通量测序技术在猪肉品质研究中的应用体系目前尚不完善,且较少找到实践案例,但近年来随着该技术的不断发展,运用高通量测序技术研究猪肉在发育过程中不同时期、不同品种间的基因差异,挖掘性状基因和调控机制,继而认识和研究肉品差异的遗传机理的方法被越来越广泛地应用。运用高通量基因组测序结合相关的基因技术,不但可以对猪的肉品质量进行分析预测,还能通过遗传育种等技术从根本上改善肉品质量,保留良好的生长性能,提高肉类加工行业的经济效益。
在遗传改良上,研究人员通过收集猪种资源,结合各种高通量测序技术进行多组学比较分析,继而挖掘肉品质关键相关基因。在此基础上建立猪的基因数据库,将对于未来猪的遗传育种研究有重要意义,发展猪的分子辅助育种体系,开展猪的基因工程,进行分子标记开发。将分子标记应用于猪肉品质相关性状的指数选择,建立高效的技术育种体系,通过建立不同标记组合的试验猪群,选择最佳标记组合的基因型个体开展定向选育。在猪基因工程技术方面,制备可控表达的转基因猪。利用体细胞转基因克隆,进一步全面解析猪肌肉和脂肪发育分子机制,精准调控猪肌肉和脂肪发育,培育优质猪种。
[1] Larzul C, Lefaucheur L, Ecolan P,et al. Phenotypic and genetic parameters for longissimus muscle fiber characteristics in relation to growth, carcass, and meat quality traits in large white pigs[J]. J Anim Sci, 1997, 75(12): 3126.
[2] Salas R C D, Mingala C N. Genetic factors affecting pork quality: Halothane and rendement napole genes[J]. Anim Biotechnol, 2016, 28(2): 148-155.
[3] Rothberg J M, Leamon J H. The development and impact of 454 sequencing[J]. Nat Biotechnol, 2008, 26(10): 1117-1124.
[4] Lizardi P M. Next generation sequencing by hybridization[J].Nat Biotechnol, 2008, 26(6): 649-650.
[5] Ondov B D, Varadarajan A, Passalacqua K D,et al. Efficient mapping of applied biosystems SOLiD sequence data to a reference genome for functional genomic applications[J].Bioinformatics, 2008, 24(23): 2776-2777.
[6] Cirulli E T, Singh A, Shianna K V,et al. RSecseraerchening the human exome: a comparison of whole genome and whole transcriptome sequencing[J]. Genome Biol, 2010, 11(5): R57.
[7] Wilhelm B T, Marguerat S, Watt S,et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution[J]. Nature, 2008 , 453: 1239-1243.
[8] Alagna F, Dagostino N, Torchia L,et al. Comparative 454 pyrosequencing of transcripts from two genotypes during fruit development[J]. BMC Genomics, 2009, 10: 399.
[9] Wang E T, Sandberg R, Luo S,et al. Alternative isoform regulation in human tissue transcriptomes[J]. Nature, 2008,456: 470-476.
[10] Patrica A. Quality of pork in Danmark pig[J]. Fanning(supplement), 1995, 10: 56-57.
[11] 谭林, 姜海龙. 肌内脂肪含量与猪肉品质的相关性分析[J].饲料博览, 2010(12): 11-13.
[12] 章 杰, 罗宗刚, 陈磊, 等. 荣昌猪和杜洛克猪肉质及营养价值的比较分析[J]. 食品科学, 2015, 36(24): 127-130.
[13] Bai H, Zhu J, Sun Y,et al. Identi fication of genes related to beak deformity of chickens using digital gene expression pro filing[J].PLoS One, 2014, 9(9): 107050.
[14] Shefer G, Wleklinski-lee M, Yablonka-reuveni Z. Skeletal muscle satellite cells can spontaneously enter an alternative mesenchymal pathway[J]. J Cell Sci, 2004, 117(22): 5393-5404.
[15] 张凤. Myostatin对猪肌卫星细胞和脂肪来源干细胞成脂分化的调控机制研究[D]. 武汉: 华中农业大学, 2016: 77-90.
[16] Ray K. NASH: KLF6 activates PPARalpha signaling in hepatic steatosis[J]. Nat Rev Gastro Hepat, 2013, 10(3): 128.
[17] Wei S, Zhang L, Zhou X,et a1. Emerging roles of zinc finger proteins in regulating adipogenesis[J]. Cell Mol life Sci, 2013,70(23): 4569-4584.
[18] 朱江江 , 林亚秋 , 左璐璐 , 等 . 牦牛 KLF5、KLF6、KLF7 基因的克隆表达及其与肌内脂肪含量的相关性分析[J]. 畜牧兽医学报, 2017, 48(3): 416-424.
[19] 邢凯, 侯卓成, 王晓铄, 等. 背膘极端表现猪脂肪和肝脏组织的表达谱分析[A]. 中国畜牧兽医学会2013年学术年会论文集[C] .北京: 中国畜牧兽医学会, 2013.
[20] Li G, Li Y, Li X,et al. MicroRNA identity and abundance in developing swine adipose tissue as determined by Solexa sequencing[J]. J Cell Biochem, 2011, 112(5): 1318-1328.
[21] 张冬杰, 刘娣, 汪亮, 等. 民猪和大白猪背最长肌差异表达基因的筛选与注释[J]. 畜牧兽医学报, 2013, 44(2): 181-187.
[22] Bai Y, Huang J M, Liu G,et al. A comprehensive microRNA expression pro file of the backfat tissue from castrated and intact full-sib pair male pigs[J]. BMC Genomics, 2014, 15(1): 47.
[23] Chen C, Deng B, Qiao M,et al. Solexa Sequencing identification of conserved and novel micrornas in backfat of large white and chinese meishan pigs[J]. PLoS One, 2012, 7(2): 31426.
[24] Warner R D, Greenwood P L, Pethick D W,et al. Genetic and environmental effects on meat quality[J]. Meat Sci, 2010, 86(1):171-183.
[25] Ohlendieck K. Proteomics of skeletal muscle glycolysis[J].BBA, 2010, 1804(11): 2089-2101.
[26] Larzul C, Lefaucheur L, Ecolan P,et al. Phenotypic and genetic parameters for longissimus muscle fiber characteristics in relation to growth, carcass, and meat quality traits in large white pigs[J]. J Anim Sci, 1997, 75(12): 3126.
[27] Kim J M, Lee S H, Ryu Y C. Comparisons of meat quality and muscle fibre characteristics on multiple pig breeds and sexes using principal component analysis[J]. Anim Prod Sci, 2017:http://doi.org/10.1071/AN16223.
[28] 彭兴. 猪肌肉发育相关microRNA-21靶标基因TSC1和SMAD7的鉴定[D]. 长沙: 湖南农业大学, 2013: 37-55.
[29] Hou X, Tang Z, Liu H, et al. Discovery of MicroRNAs associated with myogenesis by deep sequencing of serial developmental skeletal muscles in pigs[J]. PLoS One, 2012,7(12): 52123.
[30] 朱嘉宇. 猪肌纤维类型关键基因筛选及miR-423-5p的功能研究[D]. 杨凌: 西北农林科技大学, 2016: 72-73.
[31] Nielsen M, Hansen J H, Hedegaard J, et al. MicroRNA identity and abundance in porcine skeletal muscles determined by deep sequencing[J]. Anim Genet, 2010, 41(2): 159-168.
