分子印迹固相萃取- 高效液相色谱法测定牛奶中四环素类抗生素残留
2018-03-02付珍珍李增威刘书亮邹立扣敖晓琳陈姝娟
付珍珍,曾 月,李增威,何 利,周 康,刘书亮,邹立扣,敖晓琳,陈姝娟
(四川农业大学 食品学院,四川 雅安 625014)
四环素类抗生素(tetracyclines,TCs)是一类由放线菌产生的广谱抗生素,包括土霉素(oxytetracycline,OTC)、四环素(tetracycline,TC),以及金霉素(chlortetracycline,CTC)等[1-2],主要用于动物疾病的预防、治疗和促进动物生长[3]。但由于使用不规范,也造成了TCs在动物性食品中的残留[4-6],给动物性食品安全带来巨大隐患,人类若长期食入含有TCs残留的食物易造成牙齿畸形、肝脏损害等。研究表明,抗生素进入动物体内后约70%不能被吸收,不吸收的部分大都以原形或代谢产物的形式排出,污染土壤[7]。据报道,在用动物排泄物施肥的土壤的0~40 cm表层,检测到浓度高达32.3 mg·kg-1的土霉素和26.4 mg·kg-1的金霉素残留[8]。目前,检测TCs的方法主要有薄层色谱法、高效液相色谱法、液相色谱- 质谱联用法、酶联免疫分析法和微生物法等[9-10]。其中,高效液相色谱法为实验室最常用的方法。该方法成本较低,灵敏度较高,检出限可低至μg·L-1级别,被认为是目前检测TCs的首选方法[11]。
TCs以痕量存在于食品中,而食品基质组成复杂,因此需要通过有效的预处理方法来富集样品中的TCs以便于后续检测的进行[12]。预处理不当会引入新的干扰物质影响检测的准确度。目前用于TCs预处理的方法主要有液- 液萃取、固相萃取[13-14]和基质辅助分散固相萃取[15]等,其中:液- 液萃取操作烦琐,选择性不高,且使用的溶剂对环境污染较大;基质辅助分散固相萃取法不易标准化,受操作者个体差异影响较大;固相萃取因其操作简便、选择性吸附较高、平衡时间短等优点,得到了广泛应用。分子印迹技术(molecularly imprinted technology,MIT)是对目标物质进行特异性识别的一种新型技术[16-17],具有特异性强、稳定性高和可重复使用的优点,在分离、催化剂和仿生传感器等方面应用成效良好[18]。
本实验运用MIT,将功能单体甲基丙烯酸(methacrylic acid,MAA)与模板分子盐酸强力霉素(doxycycline hydrochloride,DC),通过沉淀聚合制备出对TCs有特异性吸附的分子印迹聚合物(molecularly imprinted polymers,MIPs),并结合传统固相萃取技术,将其作为填料用于样品预处理,旨在促进MIT在动物性食品抗生素残留检测领域的应用发展,并为痕量物质的富集及检测提供一定的实验基础与参考。
1 材料与方法
1.1 仪器与试剂
TGL- 16G型高速离心机、Nicolef is 10型红外光谱仪,德国赛默飞世尔科技有限公司集团;HH数显恒温水浴锅,江苏金坛市金城国胜实验仪器厂;UV- 1800PC型紫外分光光度计,上海美谱达仪器有限公司;SB- 5200DTDN型超声波清洗仪,宁波新芝生物科技股份有限公司;ZMQS5V001型Milli- Q超纯水系统,美国密理博公司;Waters 2487型高效液相色谱仪,美国Waters公司;DZF- 6050型真空干燥箱,上海三发科学仪器有限公司。
5个品牌的250 mL盒装牛奶从市场购得;MAA、偶氮二异丁腈(AIBN)、乙腈、甲醇、乙酸、草酸,成都市科龙化工试剂厂;乙二醇二甲基丙烯酸酯(EGDMA),成都化夏化学试剂有限公司;乙腈、甲醇,色谱纯,广东光华科技股份有限公司;DC,纯度95%~102%,成都化夏化学试剂有限公司;TC、OTC、CTC标准品,纯度95%,上海瑞永生物科技有限公司。除有特别说明外,所有试剂均为分析纯,实验用水均为超纯水。
1.2 实验方法
1.2.1 四环素类分子印迹聚合物的制备
取0.15 mmol DC、1.80 mmol MAA和9.00 mmol EGDMA溶于30 mL乙腈- 丙酮(体积比16∶14)溶液,超声5 min后加入12 mg AIBN,通氮气除氧10 min,密封后于恒温水浴箱中60 ℃反应24 h得到聚合物。用乙腈- 甲醇(体积比1∶1)溶液为洗液,在索氏提取器内洗脱48 h,再用甲醇- 水(体积比1∶1)洗去模板分子DC。洗脱完毕后于40 ℃真空干燥至质量恒定,得到MIPs。
采用同样方法不加模板分子制备非印迹物(NIPs)。
1.2.2 四环素类分子印迹聚合物的表征
(1)红外光谱。将样品用KBr压片法制成薄片,在500~4 000 cm-1范围内测定MIPs、NIPs、MAA、DC,以及EGDMA的红外光谱。
(2)扫描电镜。用甲醇溶液分散MIPs并涂于载玻片上,对其喷金后用XL- 30ESEM型扫描电镜观察聚合物形态特征。
1.2.3 MIPs吸附特性研究
(1)HPLC检测条件。色谱柱,Sepax HP- C18,250 mm×4.6 mm,5 μm;流动相,乙腈和0.01 mol·L-1草酸- 水(体积比8∶2)溶液;流速,1.0 mL·min-1;柱温,30 ℃;进样量,20 μL;检测波长,355 nm。
(2)标准溶液配制。准确称取10 mg TC(OTC、CTC)置于1 000 mL容量瓶,甲醇定容,配制浓度为10 μg·mL-1的TC(OTC、CTC)储备液。再用甲醇分别稀释为0.05、1.0、3.0、5.0、10.0 μg·mL-1的标准溶液系列待用[19]。
(3)MIPs对TC(OTC、CTC)的吸附动力学。分别称取10 mg MIPs和NIPs于20 mL 0.1 μg·mL-1的TC(OTC、CTC)标准液。室温下振荡反应,分别于5、15、30、60、90、120 min后4 000 r·min-1离心10 min,用HPLC- UV测定上清液浓度[20],依式(1)计算吸附量(Q,μg·mg-1)。
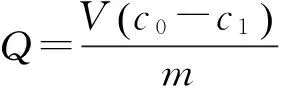
(1)
式(1)中:V为溶液的体积(mL);m为MIPs(NIPs)的质量(mg);c0为溶液初始浓度(μg·mL-1);c1为吸附残液浓度(μg·mL-1)。
(4)MIPs对TC(OTC、CTC)的静态吸附。以甲醇为溶剂,将TC(OTC、CTC)储备液配成浓度为0、0.05、0.1、0.4、0.8、1.0、2.0、4.0、6.0、8.0、10.0 μg·mL-1的标准液。分别加入2 mg MIPs,室温下振荡反应8 h,4 000 r·min-1离心10 min,取上清液过滤后用HPLC- UV检测,据式(1)计算吸附量。以同样的方式对NIPs进行考查。
1.2.4 实际样品分析
(1)MIPs固相萃取小柱制备。称取MIPs 150 mg、硅藻土75 mg,填充3.0 mL固相萃取柱,用筛板将其压紧并轻敲小柱,直至筛板与混合物紧密结合,然后用3.0 mL甲醇对MIPs固体柱进行活化。
(2)样品分析。准确量取10.0 mL牛奶,加入10.0 mL溴化四丁基铵- 乙腈(物质的量之比为1∶3)提取溶液,涡旋5 min,高速离心后取上清液,下层残渣重复提取2次。然后用20 mL乙腈除去提取液中的蛋白,再用20 mL正己烷除去脂质。50 ℃水浴蒸干后,用3 mL 0.5 mol·L-1KH2PO4溶液充分溶解后待用。以5.0 mL正己烷淋洗小柱后加入样品溶液,再用5.0 mL正己烷淋洗,弃去淋洗液,最后用5.0 mL甲醇- 冰乙酸(体积比8∶2)溶液洗脱。收集洗脱液,氮气吹干,用5.0 mL流动相溶解,过0.45 μm滤膜后进行HPLC- UV检测。
2 结果与分析
2.1 合成条件优化
2.1.1 引发剂用量
AIBN受热分解可产生引发聚合反应所需的自由基团[20]。由图1可以看出,AIBN用量会影响MIPs的吸附量,随着AIBN用量的增加,MIPs的生成量呈现先增加后减少的趋势,吸附量也相应变化,但所合成的MIPs的链长逐渐缩短,结合图1,确定AIBN最佳用量为15 mg。
2.1.2 溶剂选择与用量
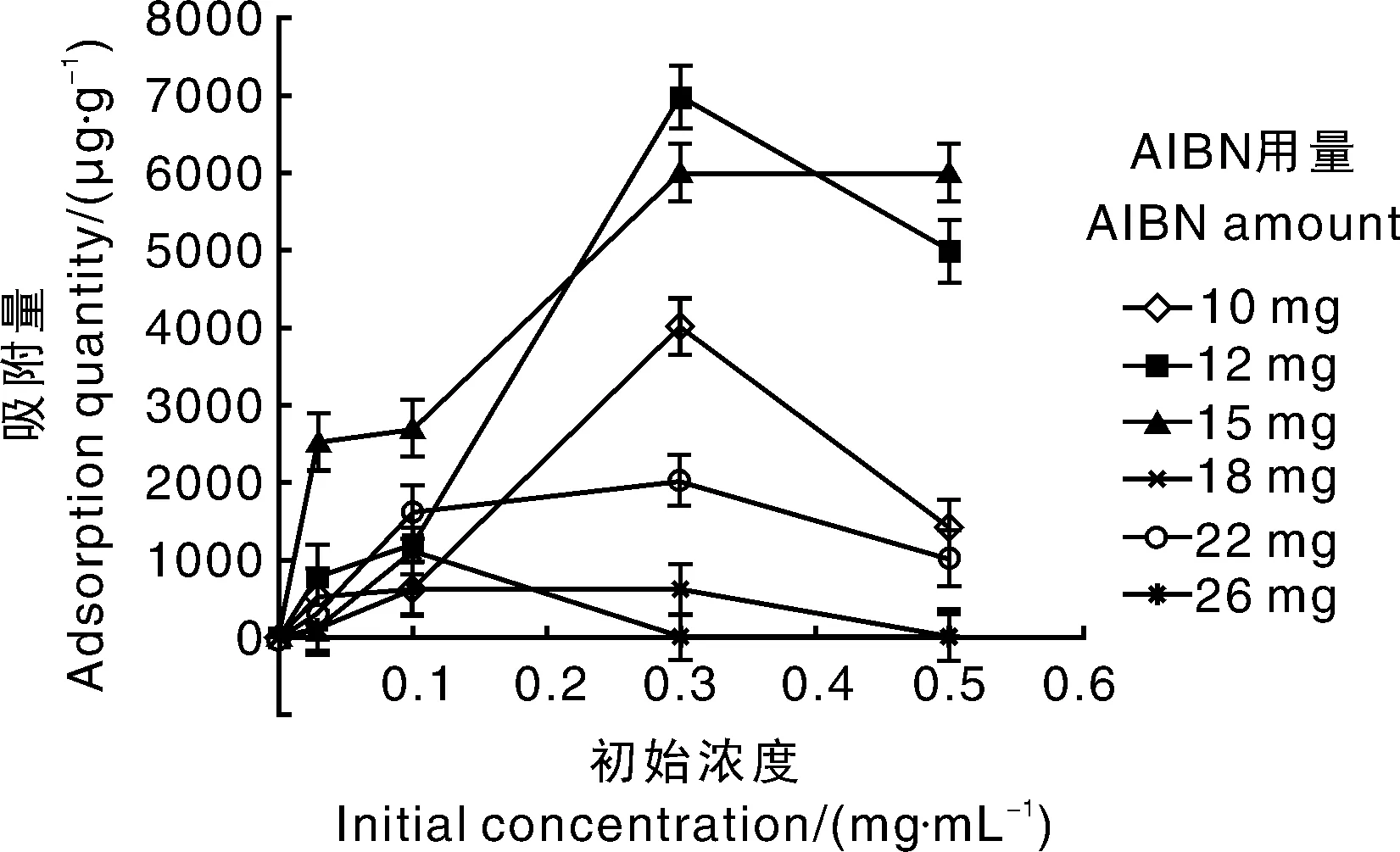
图1 AIBN用量对MIPs吸附量的影响Fig.1 Influence of AIBN amount on MIPs adsorption quantity
溶剂作为聚合反应发生的场所,不仅其种类会对MIPs产生影响,使用量也会影响MIPs的合成。由图2可看出:甲醇作为溶剂制备出的MIPs吸附量最低,这是由于甲醇极性太强,易破坏MAA与DC间的氢键;丙酮作为溶剂合成的MIPs吸附量最高,但合成速度太快,合成温度难以控制,故须与其他溶剂配合使用;乙腈作为溶剂合成的吸附性能较为稳定,对环境的耐受力大,但吸附量不够理想。将乙腈与丙酮按一定比例混合可克服各自缺点,发挥各自优点,使MIPs达到最好的吸附效果。
设计丙酮与乙腈不同体积比(10∶20,14∶16,15∶15,16∶14,20∶10)的单因素实验,来确定其最佳使用方案。结果表明,当乙腈与丙酮以16∶14的体积比混合作为溶剂时,合成的MIPs吸附量最大,且合成速度合理,条件易控制,故选用此体积比的乙腈- 丙酮混合溶液作为溶剂。
溶剂用量对MIPs生成量的影响如表1所示,用量过低,无法发生聚合反应生成MIPs,用量过高对反应体系起稀释作用,降低聚合物的生成量。故选择30 mL作为溶剂最佳用量。
2.1.3 模板分子/单体/交联剂比例
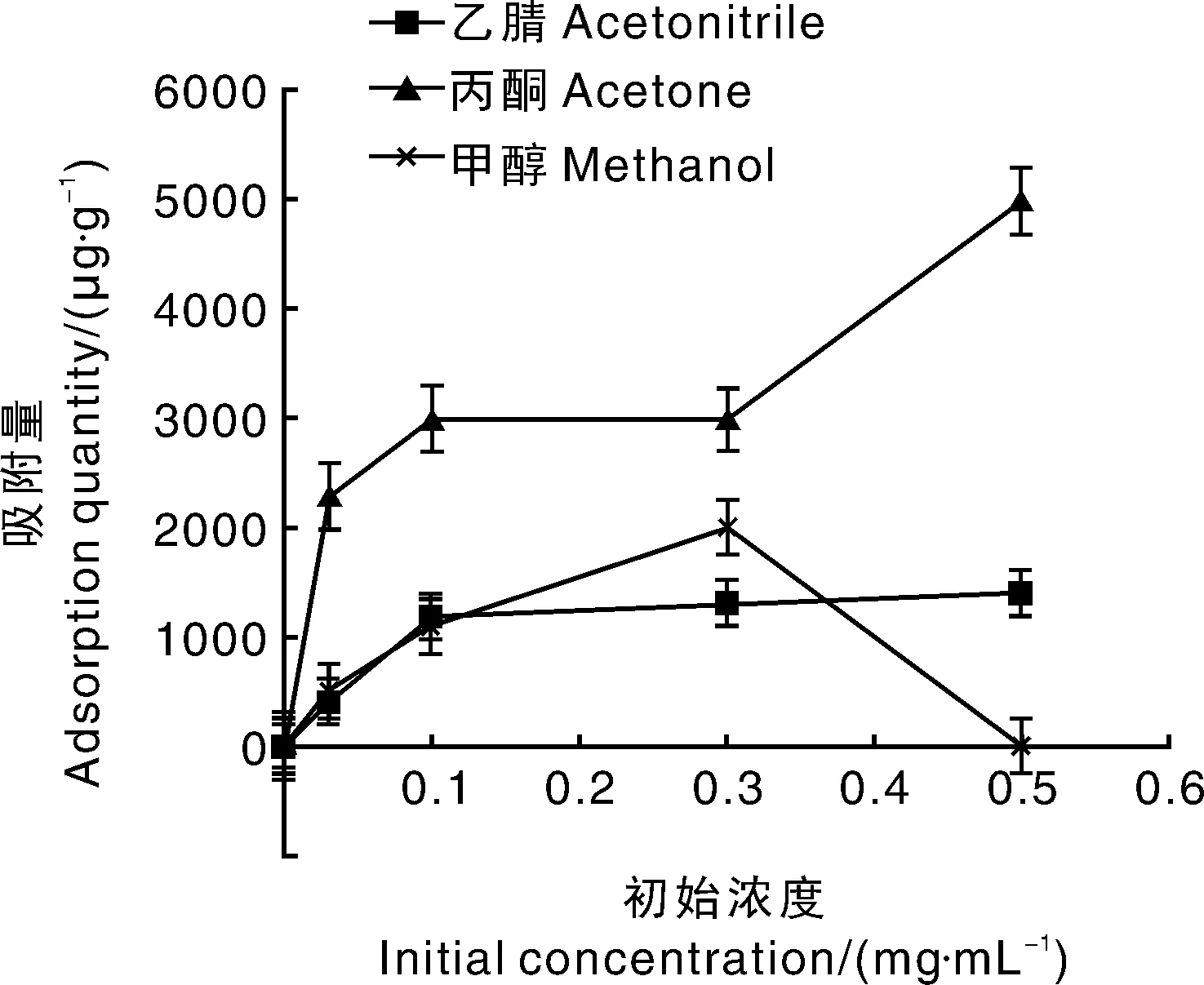
图2 溶剂对MIPs吸附量的影响Fig.2 Influence of solvent on MIPs adsorption quantity
表1溶剂用量对MIPs生成量的影响
Table1Influence of solvent dosage on MIPs quantity
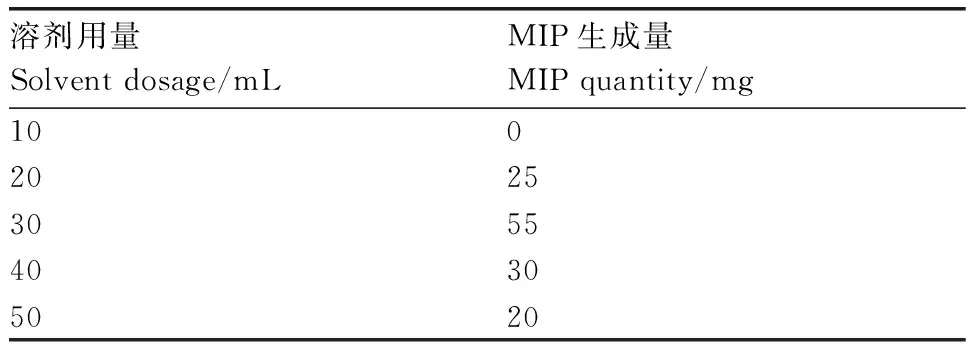
溶剂用量Solventdosage/mLMIP生成量MIPquantity/mg1002025305540305020
研究表明,模板与单体的比例(即二者物质的量之比)可对聚合物的吸附性能产生影响。比例太低难以形成完整的模板识别位置,无法实现单体与模板分子的多点结合,反之比例太高则单体过剩,会导致非特异性吸附增强[21]。本实验使用0.15 mmol DC,并保持MAA与EGDMA物质的量1∶5的比例不变,考查MAA用量对MIPs形态和合成量的影响(DC、MAA、EGDMA的物质的量之比分别为1∶1∶5、1∶4∶20、1∶6∶30、1∶8∶40、1∶10∶50、1∶12∶60、1∶14∶70)。结果表明,MIPs的聚合程度与MAA用量成正比,但随MIPs聚合程度加深,其形状由规则的球状团聚物变为不规则的团聚物,进而导致吸附量下降,因此选择DC、MAA、EGDMA物质的量之比1∶12∶60为最佳比例。
2.2 聚合物表征
2.2.1 红外光谱分析
图3为MIPs、MAA、EGDMA的红外光谱图,在1 602 cm-1位置,MIPs相比MAA、EGDMA振动幅度明显降低,有对应于C=C双键伸缩振动的弱红外吸收峰,表明此处MAA和EGDMA成功交联。在3 300 cm-1羟基的特征峰[22]发生明显振动,表明MIPs含有羟基,能与目标化合物通过氢键结合。
由图4可知,洗脱后的MIPs在2 400 cm-1和820 cm-1处较洗脱前的MIPs伸缩振动明显减小,分别为DC中芳烃C—H和N—H的特征峰,表明DC与EGDMA成功交联。洗脱后的MIPs与NIPs的红外谱图基本重合,因二者具有相同的化学组成与骨架结构,且NIPs中不含DC,表明MIPs中的DC已被完全洗脱。
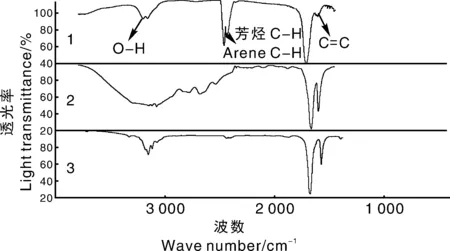
1,MIPs;2,MAA;3,EGDMA.图3 MIPs、MAA与EGDMA的红外光谱图Fig.3 FT- IR spectra of MIPs、MAA and EGDMA

1,洗脱前MIPs;2,洗脱后MIPs;3,NIPs。1, MIPs before elution; 2, MIPs after elution; 3, NIPs.图4 MIPs洗脱前后与NIPs的红外光谱图Fig.4 FT- IR spectra of MIPs before and after elution and NIPs
2.2.2 扫描电镜分析
电镜扫描结果如图5所示。MIPs表面光滑,呈团聚球状,由于与DC发生了聚合反应,表面形成与DC互补的孔穴,可有效地用于特异性识别DC。
2.2.3 吸附试验
在静态吸附实验中,MIPs表面孔穴的数量及互补度对吸附效果具有决定性的作用[22]。从图6可知,MIPs对OTC的吸附量较TC大,原因是MIPs表面的孔穴与OTC的互补度较大,吸附量也更大。吸附动力学结果如图7所示,MIPs对TC(OTC、CTC)的吸附速度初期均较后期快,可能是因为吸附初期MIPs表面的孔穴与TC(OTC、CTC)进行特异性结合导致表面的孔穴被堵塞,之后TC(OTC、CTC)渗入MIPs内部需要的时间更长。NIPs与MIPs相比吸附量更低,原因是NIPs未与DC发生聚合,没有形成与TC(OTC、CTC)互补的孔穴,因此没有特异性吸附[23]。

图5 MIPs的电镜扫描图Fig.5 SEM images of MIPs

图6 MIPs对四环素、土霉素和金霉素的静态吸附曲线Fig.6 Static sorption of TC, OTC and CTC on MIPs
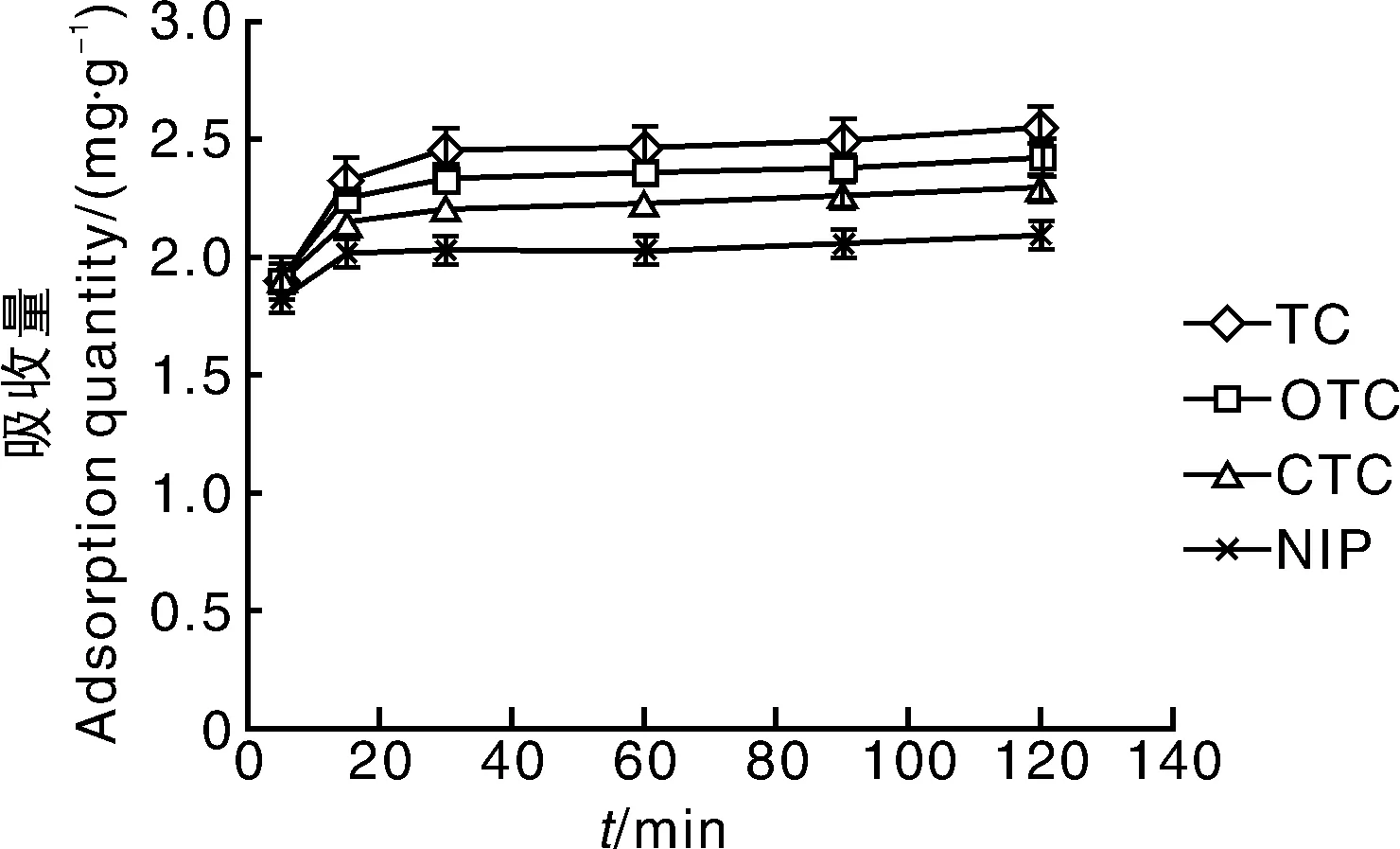
图7 MIPs对四环素、土霉素和金霉素的动态吸附曲线Fig.7 Sorption kinetics of TC, OTC and CTC on MIPs
2.3 方法性能评价
2.3.1 线性范围、检出限和相对标准偏差
图8为DC、TC、OTC和CTC混合标准工作液的色谱图,初始浓度均为0.05 mg·mL-1。在1.2.3节HPLC检测条件下重复测定标准液3次,以峰面积为纵坐标,标准液浓度为横坐标绘制标准曲线。结果如表2所示,在0.05~10.0 μg·mL-1范围内TC、OTC和CTC的浓度与吸附量呈线性关系,相关系数(R2)大于0.999 2。最低检出限和定量限分别按3倍信噪比和10倍信噪比计算,TC和OTC的检出限为0.02 μg·mL-1,定量限为0.10 μg·mL-1,CTC的检出限为0.05 μg·mL-1,定量限为0.25 μg·mL-1。此结果比现有研究报道中采用HPLC法测定TC、OTC和CTC的检出限低[24-26],相对标准偏差小于3.80%(n=3)。综上,本实验方法具有较宽的线性范围、较高的灵敏度,以及良好的重现性。
2.3.2 加标回收实验
利用本研究建立的方法测定不含TCs的牛奶样品中添加混合标准溶液后TCs的回收率。根据线性范围内最低浓度的1、2、10、20倍进行加标。表3为在空白牛奶中分别加入0.05、0.1、0.5、1.0 μg·mL-1的TC、OTC、CTC,按照1.2.4节方法处理,在1.2.3节检测条件下,重复合成产物5次后的检测结果。结果显示,MIPs对TC、OTC和CTC的平均加标回收率分别为82.0%~86.3%、82.0%~86.0%和79.4%~85.4%,对于中低浓度的加标水平回收率较高,高浓度加标水平的平均回收率与目前最新研究结果持平[27],说明本实验方法具有较高的准确性。
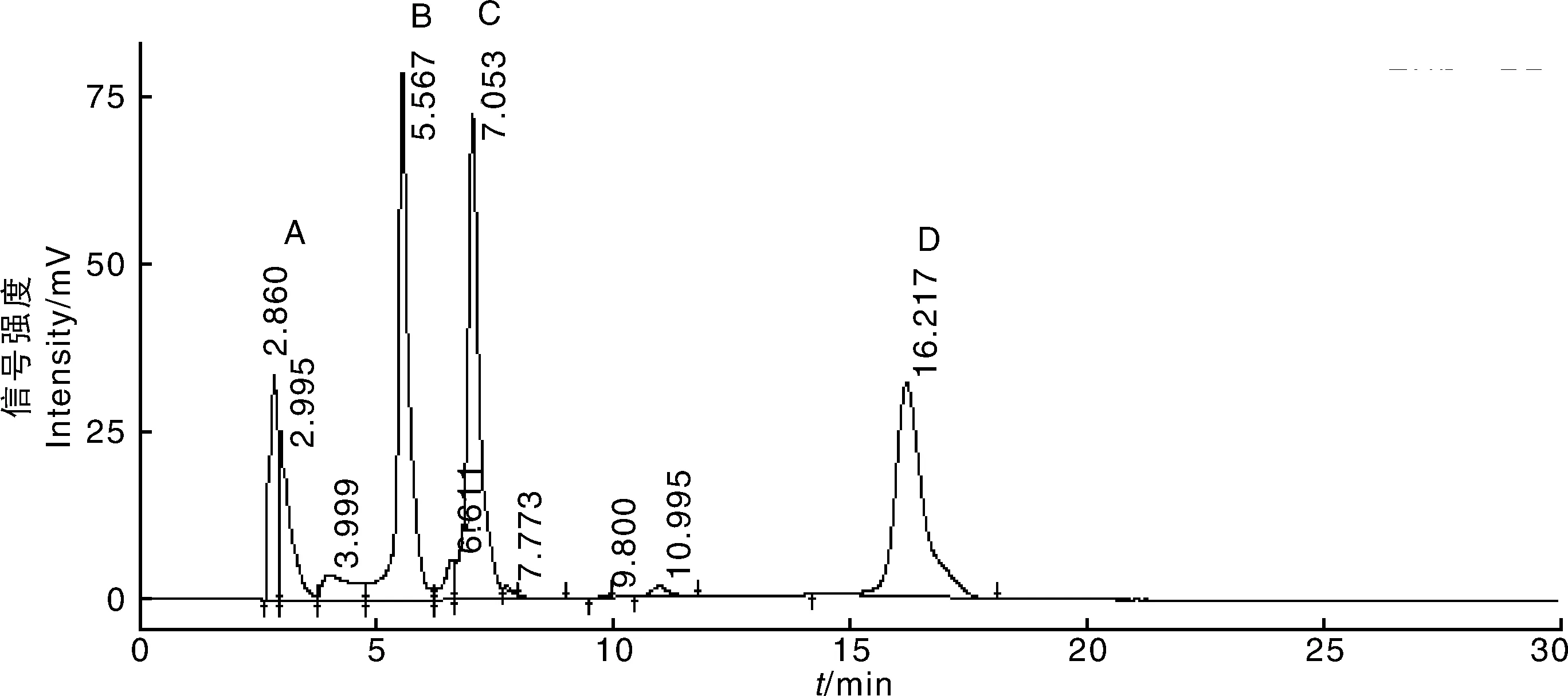
图8 DC(A)、TC(B)、OTC(C)和CTC(D)高效液相色谱图Fig.8 Chromatograms of DC(A), TC(B), OTC(C) and CTC(D)
表2四环素、土霉素和金霉素的标准曲线
Table2Standard curves of TC, OTC and CTC
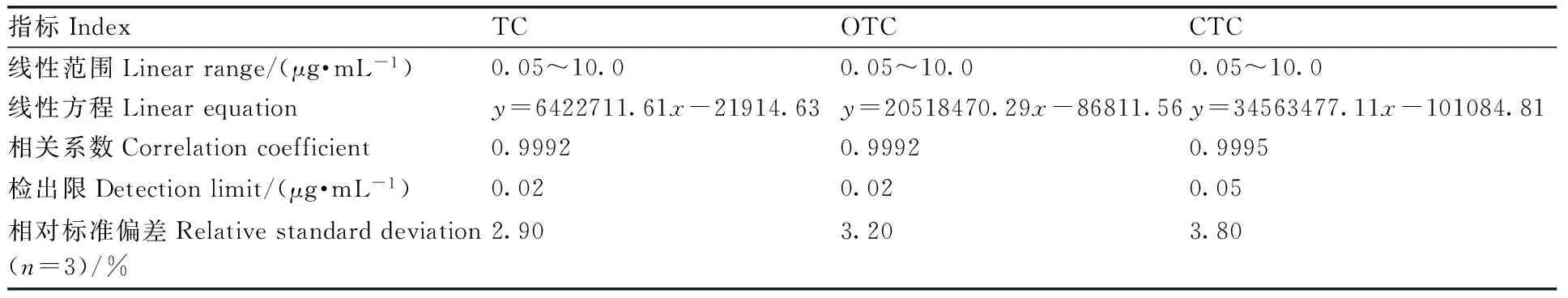
指标IndexTCOTCCTC线性范围Linearrange/(μg·mL-1)0.05~10.00.05~10.00.05~10.0线性方程Linearequationy=6422711.61x-21914.63y=20518470.29x-86811.56y=34563477.11x-101084.81相关系数Correlationcoefficient0.99920.99920.9995检出限Detectionlimit/(μg·mL-1)0.020.020.05相对标准偏差Relativestandarddeviation(n=3)/%2.903.203.80
表3TC、OTC和CTC的加标回收结果
Table3Recoveries of TC, OTC and CTC
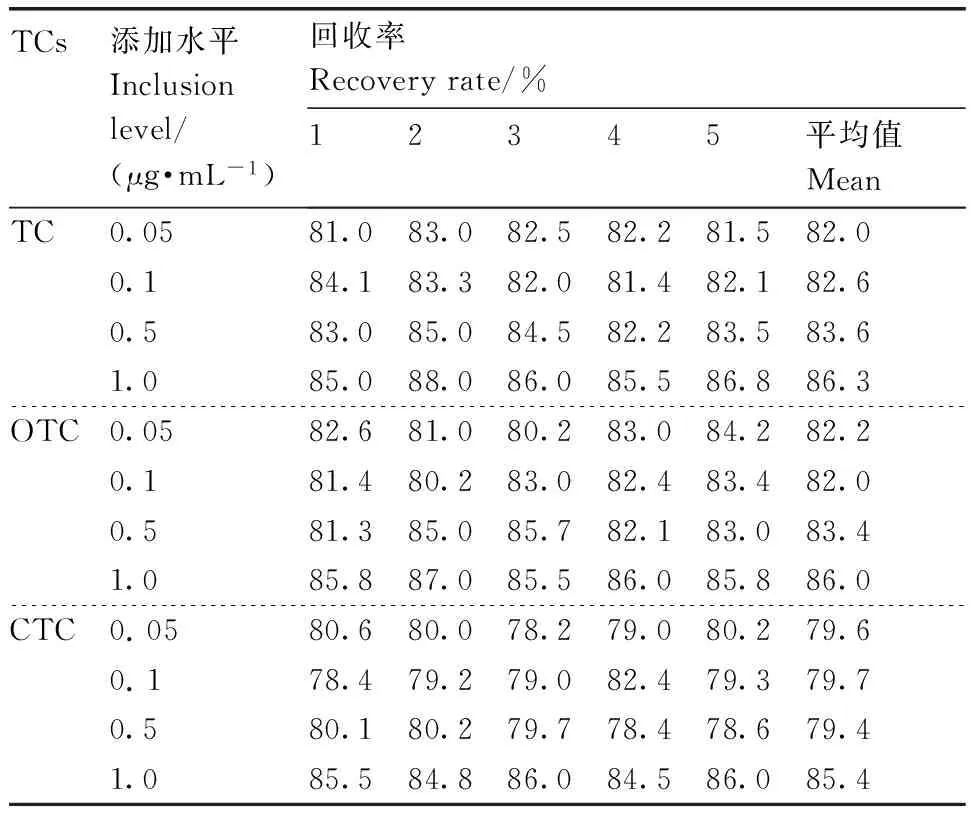
TCs添加水平Inclusionlevel/(μg·mL-1)回收率Recoveryrate/%12345平均值MeanTC0.0581.083.082.582.281.582.00.184.183.382.081.482.182.60.583.085.084.582.283.583.61.085.088.086.085.586.886.3OTC0.0582.681.080.283.084.282.20.181.480.283.082.483.482.00.581.385.085.782.183.083.41.085.887.085.586.085.886.0CTC0.0580.680.078.279.080.279.60.178.479.279.082.479.379.70.580.180.279.778.478.679.41.085.584.886.084.586.085.4
2.4 实际样品检测
选取5个品牌的市售牛奶,每个品牌分别进样3次,编号依次设定为1- 1,1- 2,1- 3,2- 1,…,5- 3。按1.2.4节方法处理,在1.2.3节所述条件检测。结果如表4所示,2个品牌检出TC,残留量分别为0.04、0.02 μg·mL-1,1个品牌检出OTC,0.03 μg·mL-1,均未超过我国农业部235号公告规定的TCs在牛奶中残留的最大限量100 μg·kg-1。
为比较MIPs固相萃取柱和传统固相萃取柱的萃取效果,分别用二者对样品进行处理。在空白牛奶中添加适量的TC、OTC和CTC标准液,将其分为等量的2份(A和B),A组按1.2.4节方法进行处理,B组用传统型的NIPs固体柱处理,于1.2.3节条件下进行检测。如图9所示,经MIPs- SPE处理的牛奶样品的TC、OTC和CTC峰面积响应值均比通过NIPs- SPE处理的样品峰面积响应值高2.4倍左右,表明MIPs- SPE能特异性地吸附牛奶中的TC、OTC和CTC,提高了检测的灵敏度及结果的准确度、精确度,有望用于土壤、动物源食品和水体等背景复杂样品的检测。而传统固相萃取柱对TCs的特异性吸附不及MIPs高,可能会将异源物质富集在所需样品中,且样品中存在的杂质会影响固相萃取柱对目标物的富集能力,所以传统固相萃取柱的萃取效果不及MIPs固相萃取柱。
表4牛奶样品检测结果
Table4Detection result of TCs in milk
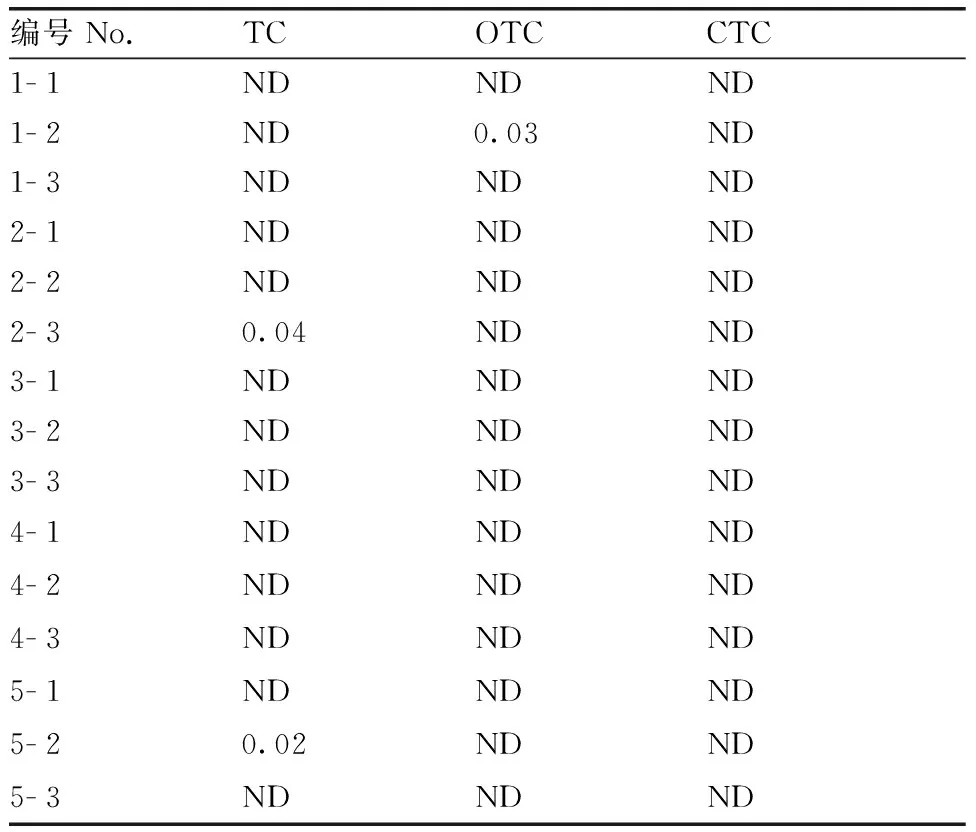
μg·mL-1
ND,未检出。
ND, Not detected.
3 小结
本文系统论述了MIPs的制备、表征,以及在实际样品中的应用。实验表明,以30 mL乙腈- 丙酮混合溶液(体积比16∶14)作为溶剂,0.15 mmol DC为模板分子,以DC/MAA/EGDMA=1∶12∶60的比例与15 mg AIBN进行沉淀聚合,得到的MIPs
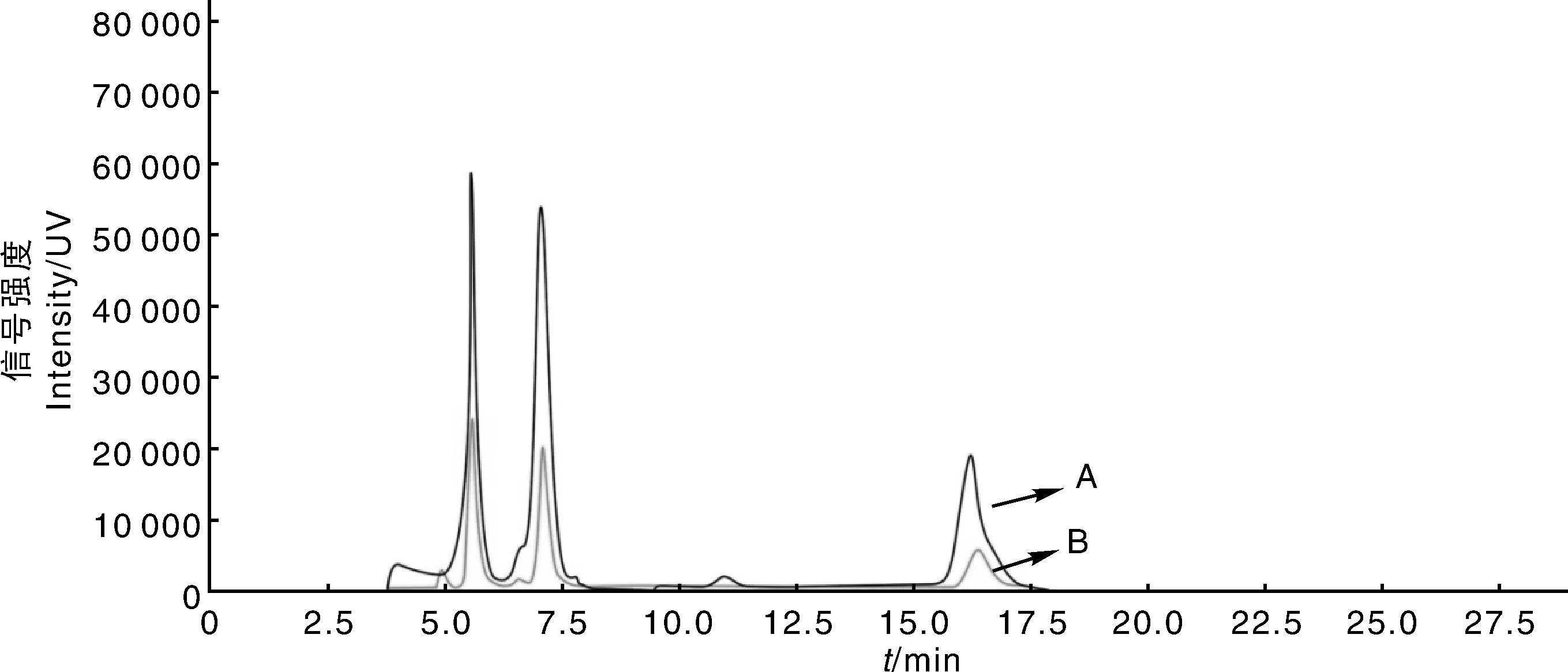
图9 不同萃取柱预处理后TCs的色谱图Fig.9 Chromatogram of TCs pretreated with different extraction column
呈规则团聚球状,且表面为蜂窝式孔状结构。吸附特性研究表明:在0.05~10.0 μg·mL-1范围内,TC、OTC和CTC的浓度与吸附量呈良好的线性关系,TC和OTC的检出限为0.02 μg·mL-1,CTC的检出限为0.05 μg·mL-1,相对标准偏差均小于3.80%。TC、OTC和CTC的平均加标回收率分别为82.0%~86.3%、82.0%~86.0%和79.4%~85.4%。将MIPs应用于复杂样品中TCs的富集,具有特异性强、操作简便的优势,可作为一种高效的TCs检测的预处理方法,用于动物源食品中低浓度TCs残留检测。
[1] 邱旭, 张健玲, 黄慧贤,等. 超高效液相色谱- 串联质谱法测定冻烤鳗中4种四环素类药物残留[J]. 福建分析测试, 2016, 25(1):46-50.
QIU X, ZHANG J L, HUANG H X, et al. Determination of 4 tetracyclines residues in frozen roasted eel tissue by UPLC- MS/MS[J].FujianAnalysis&Testing, 2016, 25(1): 46-50. (in Chinese with English abstract)
[2] 贺德春, 许振成, 吴根义. 四环素类抗生素的环境行为研究进展[J]. 动物医学进展, 2011, 32(4): 98-102.
HE D C, XU Z C, WU G Y. Progress on residues and environmental behaivor of tetracycline antibiotics[J].ProgressinVeterinaryMedicine, 2011, 32(4): 98-102. (in Chinese with English abstract)
[3] 孙霞, 王相友. 免疫传感器在牛奶抗生素残留检测中的研究进展[J]. 食品科学, 2010, 34(10): 180-183.
SUN X, WANG X Y. Research progress in detection of antibiotic residues in milk by immunosensor[J].FoodScience, 2010, 34(10): 180-183. (in Chinese with English abstract)
[4] 王萍, 司雄元, 檀华蓉, 等. 6- 氨基青霉素烷酸分子印迹固相萃取柱在牛乳青霉素药物检测中的应用[J]. 食品与发酵工业, 2016, 42(4): 183-188.
WANG P, SI X Y, TAN H R, et al. Application of 6- APA molecular imprinted solid- phase extraction column in the detection of penicillins in milk[J].FoodandFermentationIndustries, 2016, 42(4): 183-188. (in Chinese with English abstract)
[5] 国占宝, 武玉香, 田文礼, 等. 食品中四环素类残留的酶联免疫检测试剂盒的研制[J]. 食品科学, 2011, 32(2): 333-337.
GUO Z B, WU Y X, TIAN W L, et al. Development of a new ELISA kit for tetracycline residue detection in foods[J].FoodScience, 2011, 32(2): 333-337. (in Chinese with English abstract)
[6] CHEE- SANFORD J C, MACKIE R I, KOIKE S, et al. Fate and transport of antibiotic residues and antibiotic resistance genes following land application of manure waste[J].JournalofEnvironmentalQuality, 2009, 38(3): 1086-1108.
[7] 张浩, 罗义, 周启星. 四环素类抗生素生态毒性研究进展[J]. 农业环境科学学报, 2008, 27(2): 407-413.
ZHANG H, LUO Y, ZHOU Q X, Research advancement of eco- toxicity of tetracycline antibiotics[J].JournalofAgro-EnvironmentScience, 2008, 27(2): 407-413. (in Chinese with English abstract)
[8] 罗侃, 孙润泰, 孙晓红, 等. 牛奶中四环素类抗生素残留量测定及市场调查[J]. 中国卫生检验杂志, 2006, 16(9): 1077-1078.
LUO K, SUN R T, SUN X H, et al. Determination of tetracycline antibiotics residues in milk and market investigation[J].ChineseJournalofHealthLaboratoryTechnology, 2006, 16(9): 1077-1078. (in Chinese)
[9] 杨旭, 刘美娇, 林深, 等. 限进材料固相萃取- 高效液相色谱在线联用检测牛奶中四环素类抗生素残留[J]. 分析化学, 2016, 44(1): 146-151.
YANG X, LIU M J, LIN S, et al. Determination of tetracycline residues in milk by on- line coupling of restricted access material solid phase extraction with high performance liquid chromatography[J].ChineseJournalofAnalyticalChemistry, 2016, 44(1): 146-151. (in Chinese with English abstract)
[10] 闫小峰. 四环素类抗生素残留检测方法研究进展[J]. 中国兽药杂志, 2010, 44(5): 47-50.
YAN X F. Research progress of determination methods for tetracyclines antibiotic residues[J].ChineseJournalofVeterinaryDrug, 2010, 44(5): 47-50. (in Chinese with English abstract)
[11] 杨晓芳, 杨涛, 王莹, 等. 四环素类抗生素污染现状及其环境行为研究进展[J]. 环境工程, 2014, 32(2): 123-127.
YANG X F, YANG T, WANG Y, et al. Research progress in pollution status and environmental behavior of tetracycline antibiotics[J].EnvironmentalEngineering, 2014, 32(2): 123-127. (in Chinese with English abstract)
[12] SONG J, ZHANG Z H, ZHANG Y Q, et al. Ionic liquid dispersive liquid- liquid microextraction combined with high performance liquid chromatography for determination of tetracycline drugs in eggs[J].AnalyticalMethods, 2014, 6(16): 6459-6463.
[13] CARO E, MARCE R M, CORMAC P, et al. Synthesis and appLication of an oxytetracyc line imprinted polymer for the solid- phase extraction of tetracycline antibiotics[J].AnaiyticaChimicaActa, 2005(1): 81-86.
[14] 张元, 张峰,周昱, 等.食品中四环素类药物残留检测前处理及分析方法研究进展[J]. 药物分析杂志, 2016, 36(4): 565-571.
ZHANG Y, ZHANG F, ZHOU Y, et al. Research progress on pretreatment techniques and determination methods of tetracyclines residues in food[J].ChineseJournalofPharmaceuticalAnalysis, 2016, 36(4): 565-571. (in Chinese with English abstract)
[16] 张成波, 李兆周, 侯玉泽, 等. 分子印迹技术在药物残留检测中的应用[J]. 食品科学, 2014, 35(9): 323-328.
ZHANG C B, LI Z Z, HOU Y Z, et al. Application of molecular imprinting technology in the detection of drug residues[J].FoodScience, 2014, 35(9): 323-328. (in Chinese with English abstract)
[17] YU H, MU H, HU Y M. Determination of fluoroquinolones ulfonamides and tetracyclines multiresidues simultaneously inporcine tissue by MSPD and HPLC- DAD[J].JournalofPharmaceuticalAnalysis, 2012, 2(1): 76-82.
[18] 黄晓丽, 蒲家志, 陈衬心, 等. 分子印迹技术在药学中的发展及其应用前景[J]. 安徽农业科学, 2016 (3): 3-6.
HUANG X L, PU J Z, CHEN C X, et al. Application of molecular imprinting technique in pharmaceutical science and its prospect[J].JournalofAnhuiAgriculturalSciences, 2016 (3): 3-6. (in Chinese with English abstract)
[19] SUN X, HE X, ZHANG Y, et al. Determination of tetracyclines in food samples by molecularly imprinted monolithic column coupling with high performance liquid chromatography[J].Talanta, 2009, 79(3): 926-934.
[20] 李倩. 四环素类分子印迹聚合物的合成及其识别性能的研究[D]. 上海: 中国农业科学院, 2010.
LI Q. The synthesis of tetracycline molecular imprinted polymer and the evaluation of their recognition characters[D]. Shanghai: Chinese Academy of Agricultural Sciences, 2010. (in Chinese with English abstract)
[21] 蒋宇翔, 庞道林, 何强, 等. 分子印迹技术特异性识别头孢类分子的光谱研究[J]. 光谱学与光谱分析, 2011, 31(7): 1852-1856.
JIANG Y X, PANG D L, HE Q, et al. Spectroscopy study on the selectively distinguishing cefalexin with the molecular imprinted polymer[J].SpectroscopyandSpectralAnalysis, 2011, 31(7): 1852-1856. (in Chinese with English abstract)
[22] SANCHEZ- POLO M, VELO- GALA I, JESUS J. Molecular imprinted polymer to remove tetracycline from aqueous solutions[J].MicroporousandMesoporousMaterials, 2015, 203: 32-40.
[23] DIVYA M P, YUDHISHTHIR S R, RAJAN S. Synthesis and application of tetracycLine imprinted polymer[J].AnalyticalLetters, 2010, 43(6): 919-928.
[24] 庄园, 彭英, 赵永刚, 等. 分子印迹固相微萃取- 高效液相色谱法测定水和牛奶中三种四环素类药物[J]. 分析科学学报, 2014, 30(4): 451-456.
ZHUANG Y, PENG Y, ZHAO Y G, et al. Determination of tetracyclines in water and milk by solid phase microextraction based on tetracylines molecularly imprinted polymers coupled with HPLC[J]. JournalofAnalyticalScience, 2014, 30(4): 451-456. (in Chinese with English abstract)
[25] 蔡志斌, 张英, 刘丽. 固相萃取- 高效液相色谱法同时测定牛奶中3种四环素类抗生素残留量[J]. 中国卫生检验杂志, 2007, 17(2): 270-272.
CAI Z B, ZHANG Y, LIU L. Determination of 3 kinds of tetracyclines residues in m ilk by SPE- HPLC[J].ChineseJournalofHealthLaboratoryTechnology, 2007, 17(2): 270-272. (in Chinese with English abstract)
[26] 陈小燕, 牛玉玲, 朱敏, 等. 固相萃取- 高效液相色谱法测定牛奶中四环素类抗生素[J]. 中国抗生素杂志, 2017, 42(2): 129-133.
CHEN X Y, NIU Y L, ZHU M, et al. Determination of tetracycline antibiotics in milk by solid phase extraction combined with high performance liquid chromatography[J].ChineseJournalofAntibiotics, 2017, 42(2): 129-133. (in Chinese with English abstract)
[27] 王雅群, 潘道东. 磁性分子印迹固相萃取食品中的四环素类抗生素残留[J]. 食品工业科技, 2015, 36(18): 53-58.
WANG Y Q, PAN D D. Magnetic molecularly imprinted nanocompsites combined with solid phase extraction detect tetracycline antibiotic residues[J].ScienceandTechnologyofFoodIndustry, 2015, 36(18): 53-58. (in Chinese with English abstract)
