应用体外全仿生模型初步分析2 种富硒产品中硒形态及生物可给性
2018-02-09陈尚龙刘恩岐陈安徽巫永华
陈尚龙,刘恩岐,陈安徽,刘 辉,巫永华,秦 旭
(1.徐州工程学院食品(生物)工程学院,江苏 徐州 221018;2.徐州工程学院 江苏省食品资源开发与质量安全重点建设实验室,江苏 徐州 221018)
硒是人体必需微量营养元素[1],但不是人体内合成的元素,需要通过食物从外界摄入,人体内硒水平的高低主要取决于其膳食结构、食物中的含硒量及其化学形态。为了满足人体正常硒需要,适量补充一些富硒产品是一种有效途径[2-3]。之前,对富硒产品中硒分析多限于总量测定[4]。近年来,随着对微量元素研究的深入,已有很多硒形态分析研究报道[5-9]。Mei Tie等[5]利用高效液相色谱-电感耦合等离子体质谱联用技术[10-14]发现金针菇水溶性蛋白中至少含有6 种含硒蛋白,约占总可溶性硒的7.0%;李红卫等[6]利用4-氯-3,5-二硝基三氟甲苯柱前衍生,建立高效液相色谱法测定富硒米曲霉中有机硒形态的分析方法,取得了良好的效果;王欣等[7]建立了高效液相色谱-电感耦合等离子体质谱法测定富硒食品中6 种硒形态的方法,并应用其测定了富硒大米、富硒酵母、富硒茶叶、富硒灵芝孢子粉等多种含硒功能食品中的硒形态。但这些研究都局限于产品中硒的形态分析,并没有考虑到人体复杂的消化系统对其形态转化的影响。
实验动物模型可以较好的模拟人体消化和吸收的过程,但其成本高、耗时、耗力。本实验以实验室研制的富硒蛹虫草和富硒酵母为研究对象,采用体外全仿生消化(含消化酶)技术[15-17],依据化学仿生(人工胃、肠)与医学仿生(酶的作用)原理,模拟富硒产品经胃肠环境的转运机制,研究胃、肠所含的无机物、有机物及消化酶对富硒产品中硒形态和生物可给性的影响。正辛醇在结构上与人体内的碳水化合物和脂肪类似[18],实验以正辛醇为细胞膜,建立正辛醇吸收模型,模拟富硒产品中硒在人体胃、肠中的分配行为;鉴于消化管和血管间的生物膜为类脂质膜[19],实验以单层脂质体为细胞膜,建立单层脂质体吸收模型,模拟富硒产品中硒在人体胃、肠中的分配行为。采用微波消解作为前处理方式,使用微波消解-高分辨-连续光源石墨炉原子吸收光谱(high resolution-continuum source graphite furnace atomic absorption spectrophotometry,HR-CS GFAAS)法[20-24]测定各形态硒的含量,为进一步研究富硒产品的功能和硒的生物有效性提供初步参考数据。
1 材料与方法
1.1 材料与试剂
富硒蛹虫草和富硒酵母为实验室研发产品。
高峰α-淀粉酶、胃蛋白酶、胰酶、脂肪酶、猪胆粉、氨基葡萄糖盐酸盐、尿酸、黏蛋白、牛血清白蛋白、葡萄糖醛酸、尿素、硫氰酸钾、卵磷脂(均为分析纯)上海源叶生物科技有限公司;硒标准溶液(1 g/L) 国家化学试剂质检中心;浓硝酸、NH4H2PO4、Mg(NO3)2、Pd(NO3)2、NH4NO3、质量分数30% H2O2(均为优级纯),其他化学试剂均为分析纯 国药集团化学试剂有限公司。
1.2 仪器与设备
ContrAA 700高分辨-连续光源原子吸收光谱仪(配有MPE60自动进样器) 德国Analytik Jena公司;XT-9900型智能微波消解仪、XT-9800多用预处理加热仪 上海新拓微波溶样测试技术有限公司;CascadaTM实验室超纯水系统 美国Pall公司;3-30K台式高速冷冻离心机德国Sigma公司;L550台式低速离心机 湖南湘仪实验室仪器开发有限公司。
1.3 方法
1.3.1 仪器工作参数
HR-CS GFAAS测定硒工作参数:分析谱线为196.026 7 nm,载气为高纯氩气,基体改进剂为1 g/L Pd(NO3)2和0.5 g/L Mg(NO3)2混合溶液且添加体积为5 μL,进样体积为20 μL,灰化温度为1 100 ℃,原子化温度为2 200 ℃,原子化升温速率为1 500 ℃/s。
1.3.2 基体改进剂溶液的配制
用0.075 mol/L硝酸溶液溶解1.00 g NH4H2PO4后转移至100 mL容量瓶中,再用体积分数0.5%硝酸定容至刻度,配制成质量浓度为10 g/L NH4H2PO4。同理,配制成质量浓度为1 g/L Mg(NO3)2、1 g/L Pd(NO3)2、1 g/L NH4NO3、1 g/L Pd(NO3)2和0.5 g/L Mg(NO3)2混合溶液。
1.3.3 标准工作曲线的配制
用0.075 mol/L硝酸溶液将硒标准溶液(1 g/L)逐级稀释至0.60 mg/L硒标准使用液,再通过MPE自动进样器实现标准曲线质量浓度梯度为0.03、0.09、0.15、0.30、0.45、0.60 mg/L。
1.3.4 样品前处理及其硒的测定
将样品(固体为0.3~0.5 g,液体为5~10 g,精确至0.1 mg)、浓硝酸(5~10 mL)和质量分数30% H2O2(2~5 mL)一起置于微波消解罐中,在多用预处理加热仪上进行预处理0.5~1 h,温度由室温逐级上升至120 ℃。再加入2 mL质量分数30% H2O2,按表1中步骤进行微波消解,结束后将溶液转移至50 mL烧杯中加热至近干,用0.075 mol/L硝酸溶液多次冲洗烧杯中的样液,并将冲洗液转移至25 mL容量瓶中,用0.075 mol/L硝酸溶液定容至刻度,摇匀备用,同时以同样方法做空白对照[25]。根据硒浓度的高低,用0.075 mol/L硝酸溶液对其进行适当倍数的稀释,然后再用HR-CS GFAAS进行测定。

表1 微波消解条件Table1 Microwave digestion conditions
1.3.5 胃、肠全仿生消化液的制备

表2 唾液、胃液、十二指肠液、胆汁组成成分[19,26]Table2 Ingredients of simulated saliva, gastric juice,duodenal juice and bile
根据表2,分别加入唾液、胃液、十二指肠液和胆汁中所含的有机物和无机物,利用HCl和NaHCO3调节pH值,再用超纯水定容至500 mL,置于4 ℃冷藏保存备用,使用前在此基础上分别加入相应的消化酶。
1.3.6 胃全仿生提取液的制备
准确称取1.9~2.1 g(精确至1 mg)富硒产品(富硒蛹虫草或富硒酵母)置于250 mL三角瓶中,加入5.00 mL唾液,在37 ℃条件下恒温振荡5 min后(振荡速率为60 r/min),再加入75.0 mL胃液,在37 ℃条件下恒温振荡2 h(振荡速率为60 r/min),结束后离心20 min(离心速率为4 200 r/min)达到固液分离,得上清液(胃全仿生提取液),并称质量,置于4 ℃冷藏保存备用。
1.3.7 肠全仿生提取液的制备
准确称取1.9~2.1 g(精确至1 mg)富硒产品(富硒蛹虫草或富硒酵母)置于250 mL三角瓶中,加入5.00 mL唾液,在37 ℃条件下恒温振荡5 min后(振荡速率为60 r/min),加入75.0 mL胃液,在37 ℃条件下恒温振荡2 h(振荡速率为60 r/min),再加入100.0 mL十二指肠液和40.0 mL胆汁,在37 ℃条件下恒温振荡7 h(振荡速率为60 r/min),结束后离心20 min(离心速率为4 200 r/min)达到固液分离,得上清液(肠全仿生提取液),并称质量,置于4 ℃冷藏保存备用。
1.3.8 可溶态与悬浮态的分离和测定
准确称取40 g(精确至0.01 g)全仿生提取液置于50 mL离心管中,离心10 min(离心速率为11 000 r/min)后取上清液,再过0.45 μm滤膜,得可溶态溶液[27],并称量质量。按1.3.4节方法对可溶态溶液进行消解和测定,测定结果为可溶态硒含量,再将剩余可溶态溶液置于4 ℃冷藏保存备用。悬浮态硒含量=硒总量-可溶态硒含量。
1.3.9 可溶态中有机态和无机态的分离和测定
准确称取10 g(精确至0.01 g)可溶态溶液,过D101大孔吸附树脂(用2 倍左右体积的无水乙醇浸泡24 h左右,去离子水冲至中性),用0.15 mol/L硝酸溶液以2.0 mL/min的流速洗涤树脂,收集250 mL洗出液,按1.3.4节方法对洗出液进行消解和测定[27],测定结果为可溶态中无机态硒含量。可溶态中有机态硒含量=可溶态硒含量-可溶态中无机态硒含量。
1.3.10 有机态中蛋白结合态的分离和测定
准确移取2.00 mL可溶态溶液置于50 mL离心管中,再加入18.00 mL丙酮,混合均匀后离心10 min(离心速率为6 000 r/min),去除上清液,按1.3.4节方法对所得沉淀进行消解和测定[27],测定结果为有机态中蛋白结合态硒含量。
1.3.11 有机态中多糖结合态的分离和测定
准确移取2.00 mL可溶态溶液置于50 mL离心管中,再加入14.00 mL无水乙醇,混合均匀后离心10 min(离心速率为6 000 r/min),去除上清液,按1.3.4节方法对所得沉淀进行消解和测定[27],测定结果为有机态中多糖结合态硒含量。
1.3.12 正辛醇吸收模型制备及其体外吸收
取适量正辛醇与超纯水以体积比1∶10进行混合,强力振荡均匀后转移至分液漏斗中,避光静置12 h后待其分层,上层为被水饱和的正辛醇,下层为被正辛醇饱和的水,避光保存,备用。
准确移取1.00 mL可溶态溶液置于50 mL离心管中,加入1.00 mL被水饱和的正辛醇和20.00 mL被正辛醇饱和的水,在37 ℃条件下恒温振荡5 h(振荡速率为250 r/min)后,在4 ℃条件下恒温离心10 min(离心速率为11 000 r/min),结束后用胶头滴管吸去离心管上层正辛醇相,最后用超纯水将剩余溶液定容至25 mL,按1.3.4节方法对上述溶液进行消解和测定,测定结果为正辛醇吸收模型中水溶态硒含量。正辛醇醇溶态硒含量=可溶态硒含量-正辛醇吸收模型中水溶态硒含量。
正辛醇吸收模型中分配系数Kow值为正辛醇吸收模型中醇溶态硒含量和水溶态硒含量的比值[28]。
1.3.13 单层脂质体吸收模型制备及其体外吸收
准确称取0.001 g(精确至0.1 mg)蛋黄卵磷脂溶于10.00 mL氯仿,溶解后将其转移至具塞圆底烧瓶中,然后置于37 ℃真空旋转蒸发,直至形成均匀的多层脂质体膜。
准确移取5.00 mL可溶态溶液置于50 mL容量瓶中,用超纯水定容至刻度后,全部转移至上述圆底烧瓶中,充氮气后盖好涂有凡士林的塞子,在37 ℃条件下恒温振荡1 h(振荡速率为100 r/min),使脂质体膜全部进入溶液中。然后将全部溶液转移至100 mL塑料管中,在超低温冰箱(-80 ℃)中冷冻20 min后取出,置于37 ℃水浴中融化,重复冻融3 次,促进可溶态在单层脂质体-水体系中的分配。由于单层脂质体粒径为0.3~0.35 μm,用0.22 μm滤膜抽滤上述冻融液,收集滤液[19],按1.3.4节方法对滤液进行消解和测定,测定结果为单层脂质体吸收模型中水溶态硒含量。单层脂质体亲和态硒含量=可溶态硒含量-单层脂质体吸收模型中水溶态硒含量。
单层脂质体吸收模型中分配系数DMW值为单层脂质体亲和态硒含量和单层脂质体吸收模型中水溶态硒含量的比值。
1.3.14 硒生物可给性的计算
富硒产品胃、肠全仿生提取液中硒生物可给性按下式计算:

式中:TK为富硒产品胃、肠全仿生提取液中可溶态硒含量/(μg/g);TZ为富硒产品中硒总量/(μg/g)。
2 结果与分析
2.1 分析谱线的选择
在HR-CS AAS(采用高压短弧Xe灯)分析中,选择196.026 7 nm作为硒分析谱线时,附近有1 个强吸收峰和3 个弱吸附峰。为了选择适合硒分析的谱线,实验比较了质量浓度为0.03、0.09、0.15、0.30、0.45、0.60 mg/L这6 种标样在波长196.026 7 nm附近的吸附光谱图,如图1所示。
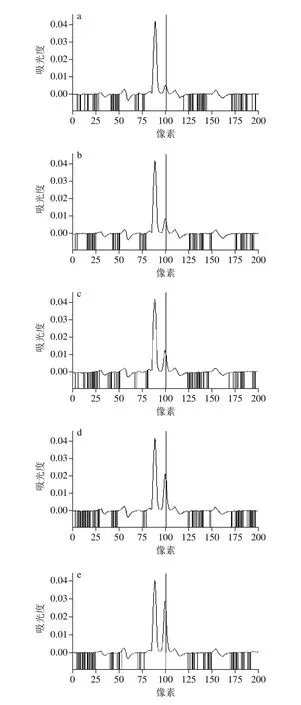
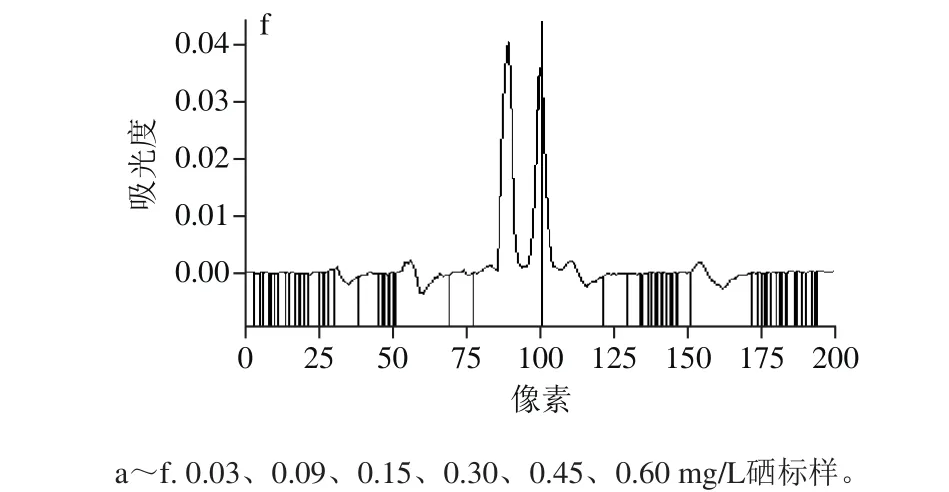
图1 波长196.026 7 nm附近吸收光谱图Fig. 1 Absorption spectra at 196.026 7 nm wavelength
由图1可知,波长196.026 7nm处吸附峰的峰高随硒质量浓度的增加而增加,而附近其他4 个吸附峰的峰高随硒质量浓度的增加基本不变,因此可以判断这4 个吸附峰为干扰峰,不能作为硒的分析谱线,HRCS AAS的分光系统是由1 个棱镜和1 个中阶梯光栅组成的高分辨单色器,通过此单色器可以获得较高的分辨率(最高可达到0.002 nm),完全可以将196.026 7 nm处吸附峰与附近其他4 个吸附峰分开。因此,实验选择196.026 7 nm作为硒分析谱线,通过HR-CS AAS的分光系统完全可以消除196.026 7 nm附近其他4 种干扰峰对硒测定的影响。
2.2 基体改进剂种类的选择
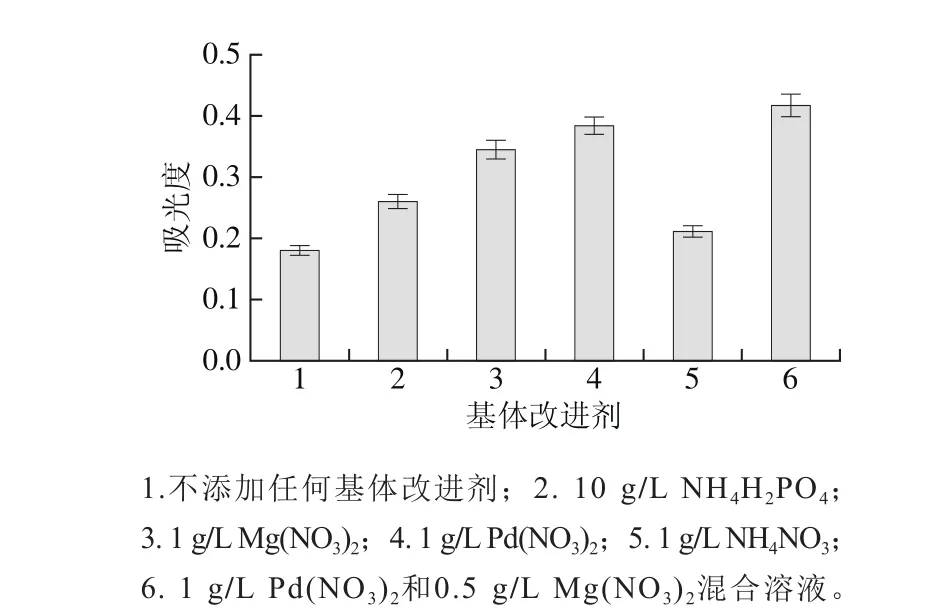
图2 基体改进剂对吸光度的影响Fig. 2 Effect of matrix modifiers on absorbance
在石墨炉原子吸收光谱法测定硒过程中,添加适合的基体改进剂可以使待测样品中基体更容易挥发,同时可以防止硒在灰化过程中挥发。实验选择NH4H2PO4、Mg(NO3)2、Pd(NO3)2、NH4NO3、Pd(NO3)2和Mg(NO3)2混合溶液作为基体改进剂[29-30],研究其对吸光度和吸收峰峰形的影响,如图2所示。以1 g/L NH4NO3作为基体改进剂,硒的吸光度增加不明显;而添加其他4 种基体改进剂,硒的吸光度增加明显,特别是1 g/L Pd(NO3)2和0.5 g/L Mg(NO3)2混合溶液,以此混合溶液作为基体改进剂不仅能提高硒的吸光度,而且还能改善硒的吸附峰峰形。因此,实验选择1 g/L Pd(NO3)2和0.5 g/L Mg(NO3)2混合溶液作为测定硒的基体改进剂。
2.3 基体改进剂添加体积的优化
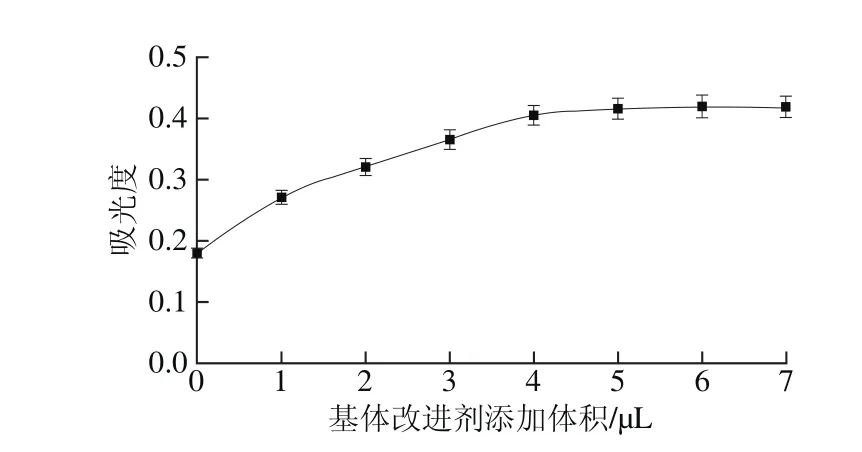
图3 基体改进剂的添加体积对吸光度的影响Fig. 3 Effect of matrix modifier volume on absorbance
基体改进剂的添加体积是影响硒吸光度的因素之一,为了最大限度地消除基体干扰,提高硒的吸光度。实验在固定其他参数条件下,只改变1 g/L Pd(NO3)2和0.5 g/L Mg(NO3)2混合溶液的添加体积,测定硒的吸光度,结果如图3所示。以1 g/L Pd(NO3)2和0.5 g/L Mg(NO3)2混合溶液为基体改进剂能显著地提高硒的吸光度,这是由于不添加基体改进剂时,过高的灰化温度会导致硒挥发损失,使硒的吸光度降低。当添加体积小于5 μL时,硒的吸光度随着基体改进剂添加体积的增大而增大;当体积不小于5 μL时,增加趋势趋于平缓。因此,实验选择添加1 g/L Pd(NO3)2和0.5 g/L Mg(NO3)2混合溶液的最佳体积为5 μL。
2.4 灰化温度的优化
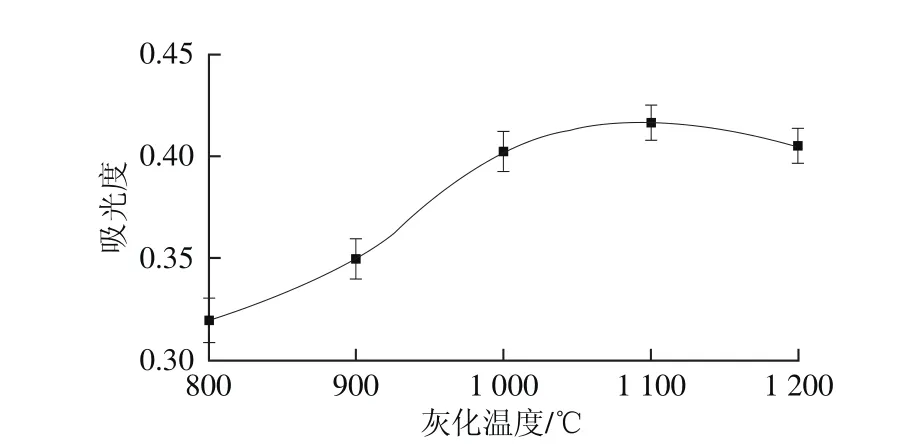
图4 灰化温度对吸光度的影响Fig. 4 Effect of aching temperature on absorbance
灰化温度是影响硒吸光度的因素之一,过低的灰化温度无法使基体完全灰化,导致背景吸光度升高;过高的灰化温度会导致硒挥发损失,使硒的吸光度降低,重复性变差。实验在固定其他参数条件下,只改变灰化温度,测定硒的吸光度,结果如图4所示。当灰化温度小于1 100 ℃时,硒的吸光度随着灰化温度的增大而增大;当灰化温度不小于1 100 ℃时,硒的吸光度开始下降。因此,实验选择最佳灰化温度为1 100 ℃。
2.5 原子化温度的优化
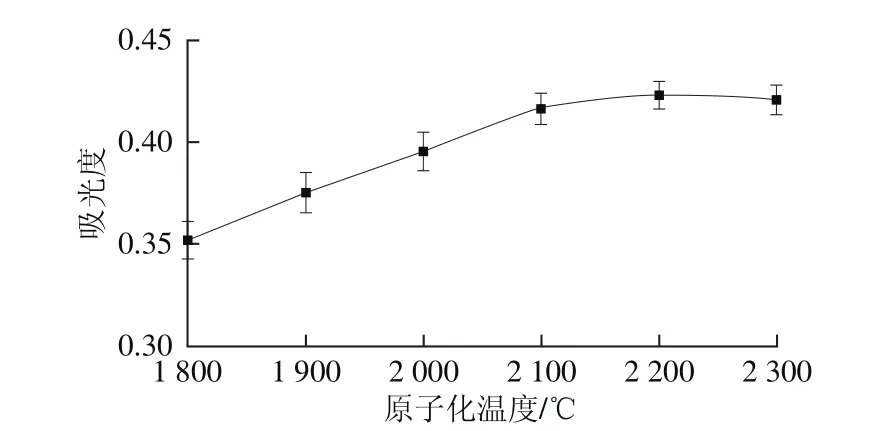
图5 原子化温度对吸光度的影响Fig. 5 Effect of atomization temperature on absorbance
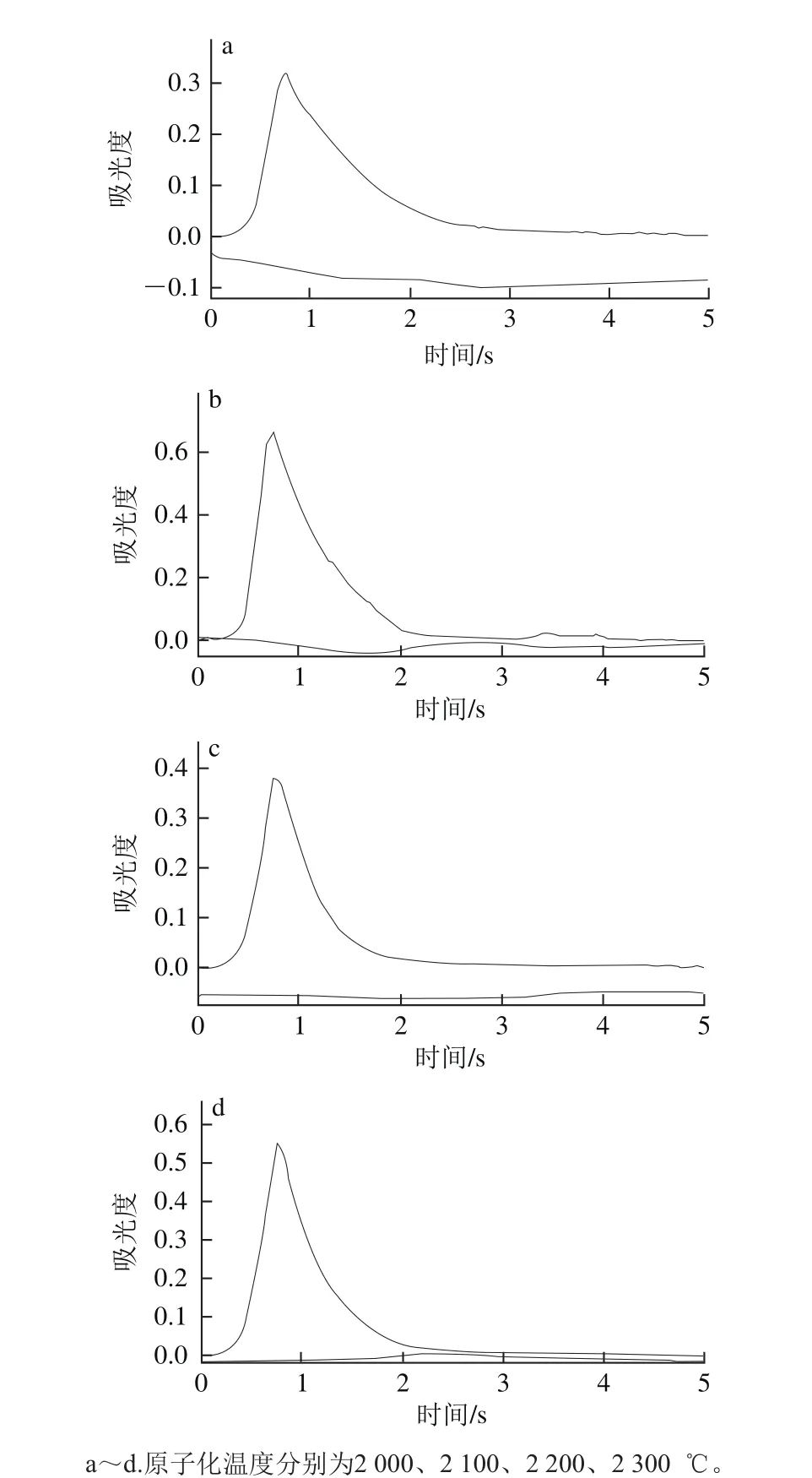
图6 原子化温度对硒吸收峰的影响Fig. 6 Effect of atomization temperature on selenium absorption peak
原子化温度也是影响硒吸光度的因素之一,过低的原子化温度无法使硒彻底原子化,导致其吸光度降低,吸附峰峰形变差;过高的原子化温度会减少石墨管的使用次数。实验在固定其他参数条件下,只改变原子化温度,测定硒的吸光度,如图5所示。不同原子化温度条件下,硒的吸收峰峰形如图6所示。由图5可知,当原子化温度小于2 200 ℃时,硒的吸光度随着原子化温度的升高而增大;当原子化温度不小于2 200 ℃时,硒的吸光度趋于稳定。由图6可知,当原子化温度为2 000 ℃和2 100 ℃时,硒的吸收峰峰形较差,拖尾现象明显;当原子化温度为2 200 ℃和2 300 ℃时,硒的吸收峰峰形较好,无明显拖尾现象。因此,实验选择最佳原子化温度为2 200 ℃。
2.6 标准工作曲线
通过ASpect CS软件设置硒的检测方法,将0.60 mg/L硒标准使用液、1 g/L Pd(NO3)2和0.5 g/L Mg(NO3)2混合溶液、空白溶液和待测样品溶液放入MPE自动进样器的对应位置,建立相应的测定序列,使用HR-CS GFAAS进行顺序测定。以硒的质量浓度(c)为横坐标、硒的吸光度(A)为纵坐标,经ASpect CS软件绘制非线性标准工作曲线,所得方程为A=(0.037 312 5+0.917 625 5c)/(1+0.304 683 9c),相关系数为0.999 2,特征质量浓度为0.004 8 mg/L,表明在0~0.60 mg/L范围内,硒的质量浓度与吸光度呈现良好的关系。
2.7 富硒蛹虫草胃、肠全仿生提取液中硒的形态分析及生物可给性
按1.3.4~1.3.13节方法对富硒蛹虫草进行处理,根据硒的标准工作曲线计算出各形态硒含量,结果见表3。
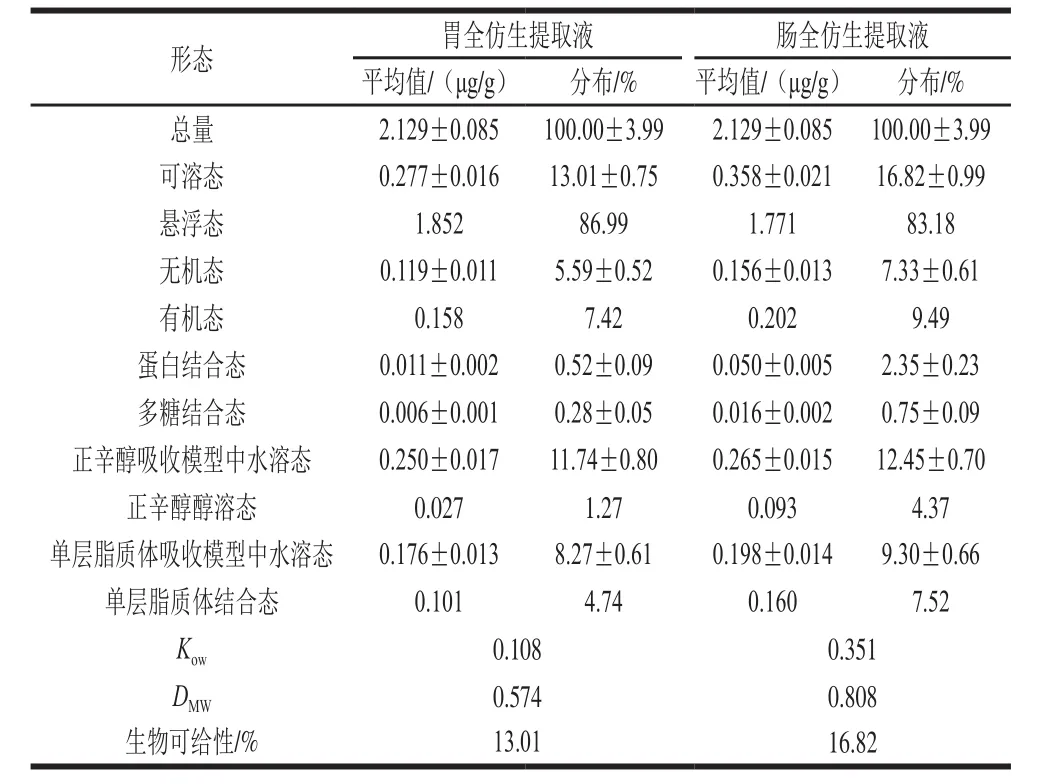
表3 富硒蛹虫草胃、肠全仿生提取液中各形态硒含量及生物可给性Table3 Speciation analysis and bioavailability assessment of Se in the gastric and intestinal digests of selenium-enriched Cordyceps militaris
由表3可知,富硒蛹虫草含有丰富的硒,达到2.129 μg/g;在胃全仿生提取液中可溶态硒为0.277 μg/g,其中有机态硒占总量的7.42%,蛋白结合态硒和多糖结合态硒分别占总量的0.52%和0.28%,正辛醇醇溶态硒和单层脂质体结合态硒分别占总量的1.27%和4.74%,Kow值和DMW值分别为0.108和0.574,硒的生物可给性为13.01%;在肠全仿生提取液中可溶态硒为0.358 μg/g,其中有机态硒占总量的9.49%,蛋白结合态硒和多糖结合态硒分别占总量的2.35%和0.75%,正辛醇醇溶态硒和单层脂质体结合态硒分别占总量的4.37%和7.52%,Kow值和DMW值分别为0.351和0.808,硒的生物可给性为16.82%。
除了悬浮态硒,富硒蛹虫草肠全仿生提取液中可溶态硒、无机态硒、有机态硒、蛋白结合态硒、多糖结合态硒、正辛醇吸收模型中水溶态硒、正辛醇醇溶态硒、单层脂质体吸收模型中水溶态硒、单层脂质体结合态硒的含量都高于胃全仿生提取液,这说明在肠消化阶段一部分悬浮态富硒蛹虫草被消化,转化成可溶态,导致可溶态硒含量升高,生物可给性变大。从各形态硒含量、Kow值和DMW值的变化分析,悬浮态硒在消化过程中转化成各形态硒,并不是仅转化成有机态硒,在肠消化过程中产生的正辛醇醇溶态硒和单层脂质体结合态硒的含量都高于各自水溶态硒,导致Kow值和DMW值变大,Kow值和DMW值越大,说明亲脂性硒含量越高。
2.8 富硒酵母胃、肠全仿生提取液中硒的形态分析及生物可给性
按1.3.4~1.3.13节方法对富硒酵母进行处理,根据硒的标准工作曲线计算出各形态硒含量,计算结果见表4。
由表4可知,富硒酵母含有丰富的硒,高达27.75 μg/g;在胃全仿生提取液中可溶态硒为2.55 μg/g,其中有机态硒占总量的6.74%,蛋白结合态硒和多糖结合态硒分别占总量的0.43%和0.25%,正辛醇醇溶态硒和单层脂质体结合态硒分别占总量的3.28%和3.57%,Kow值和DMW值分别为0.555和0.635,硒的生物可给性为9.19%;在肠全仿生提取液中可溶态硒为3.53 μg/g,其中有机态硒占总量的7.86%,蛋白结合态硒和多糖结合态硒分别占总量的1.80%和0.68%,正辛醇醇溶态硒和单层脂质体结合态硒分别占总量的6.52%和6.81%,Kow值和DMW值分别为1.05和1.15,硒的生物可给性为12.72%。
除了悬浮态硒,富硒酵母肠全仿生提取液中可溶态硒、无机态硒、有机态硒、蛋白结合态硒、多糖结合态硒、正辛醇吸收模型中水溶态硒、正辛醇醇溶态硒、单层脂质体吸收模型中水溶态硒、单层脂质体结合态硒的含量都高于胃全仿生提取液,这说明在肠消化阶段一部分悬浮态富硒酵母被消化,转化成可溶态,导致可溶态硒含量升高,生物可给性变大。从各形态硒含量、Kow值和DMW值的变化分析,悬浮态硒在消化过程中转化成各形态硒,并不是仅转化成有机态硒,在肠消化过程中产生的正辛醇醇溶态硒和单层脂质体结合态硒的含量都高于各自水溶态硒,导致Kow值和DMW值变大,Kow值和DMW值越大,说明亲脂性硒含量越高。
3 结 论
通过单因素试验,确定最佳测定条件:分析谱线为196.0267 nm,基体改进剂为1 g/L Pd(NO3)2和0.5 g/L Mg(NO3)2混合溶液,添加体积为5 μL,灰化温度为1 100 ℃,原子化温度为2 200 ℃。在此条件下,测定富硒蛹虫草和富硒酵母中总硒分别为2.129 μg/g和27.75 μg/g。除了悬浮态硒,肠全仿生提取液中其他各形态硒的含量都高于胃全仿生提取液,这说明在肠消化阶段一部分悬浮态被消化,转化成可溶态,导致可溶态硒含量升高,生物可给性变大。从各形态硒含量、Kow值和DMW值的变化分析,悬浮态硒在消化过程中转化成各形态硒,并不是仅转化成有机态硒,在肠消化过程中产生的正辛醇醇溶态硒和单层脂质体结合态硒的含量都高于各自水溶态硒,导致Kow值和DMW值变大,Kow值和DMW值越大,说明亲脂性硒含量越高。
[1] 王明洋, 方勇, 裴斐, 等. 硒对杏鲍菇营养品质和抗氧化酶活性的影响[J]. 食品科学, 2016, 37(11): 208-213. DOI:10.7506/spkx1002-6630-201611036.
[2] BODNAR M, KONIECZKA P. Evaluation of candidate reference material obtained from selenium-enriched sprouts for the purpose of selenium speciation analysis[J]. LWT-Food Science and Technology,2016, 70: 286-295. DOI:10.1016/j.lwt.2016.02.016.
[3] MAHN A, ZAMORANO M, BARRIENTOS H, et al. Optimization of a process to obtain selenium-enriched freeze-dried broccoli with high antioxidant properties[J]. LWT-Food Science and Technology, 2012,47(2): 267-273. DOI:10.1016/j.lwt.2012.01.017.
[4] 徐文军. 顺序注射氢化物发生-原子荧光光谱法测定富硒鸡蛋中的硒[J]. 食品科学, 2007, 28(8): 318-321. DOI:10.3321/j.issn:1002-6630.2007.08.076.
[5] MEI T, LI B, SUN T, et al. HPLC-ICP-MS speciation of selenium in Se-cultivated Flammulina velutipes[J]. Arabian Journal of Chemistry,2017. DOI:10.1016/j.arabjc.2017.05.012.
[6] 李红卫, 王开萍, 吴娱, 等. 柱前衍生-反相高效液相色谱法检测富硒米曲霉中有机硒形态[J]. 食品科学, 2015, 36(22): 137-141.DOI:10.7506/spkx1002-6630-201522025.
[7] 王欣, 幸苑娜, 陈泽勇, 等. 高效液相色谱-电感耦合等离子体质谱法检测富硒食品中6 种硒形态[J]. 分析化学, 2013, 41(11): 1669-1674.DOI:10.3724/SP.J.1096.2013.30491.
[8] DONG J Z, DING J, YU P Z, et al. Composition and distribution of the main active components in selenium-enriched fruit bodies of Cordyceps militaris link[J]. Food Chemistry, 2013, 137(1/2/3/4): 164-167. DOI:10.1016/j.foodchem.2012.10.021.
[9] ZHANG B, ZHOU K, ZHANG J, et al. Accumulation and species distribution of selenium in Se-enriched bacterial cells of the Bifidobacterium animalis 01[J]. Food Chemistry, 2009, 115(2): 727-734. DOI:10.1016/j.foodchem.2008.12.006.
[10] BARBOSA U A, PEÑA-VAZQUEZ E, BARCIELA-ALONSO M C, et al. Simultaneous determination and speciation analysis of arsenic and chromium in iron supplements used for iron-deficiency anemia treatment by HPLC-ICP-MS[J]. Talanta, 2017, 170: 523-529.DOI:10.1016/j.talanta.2017.04.034.
[11] JAGTAP R, MAHER W, KRIKOWA F, et al. Measurement of selenomethionine and selenocysteine in fish tissues using HPLC-ICPMS[J]. Microchemical Journal, 2016, 128: 248-257. DOI:10.1016/j.microc.2016.04.021.
[12] MARCINKOWSKA M, KOMOROWICZ I, BARAŁKIEWICZ D.Study on multielemental speciation analysis of Cr(VI), As(III) and As(V) in water by advanced hyphenated technique HPLC/ICP-DRCMS. Fast and reliable procedures[J]. Talanta, 2015, 144: 233-240.DOI:10.1016/j.talanta.2015.04.087.
[13] 金鹏飞, 吴学军, 邹定, 等. HPLC-ICP-MS研究炮制对中药砷形态的影响[J]. 光谱学与光谱分析, 2011, 31(3): 816-819. DOI:10.3964/j.is sn.1000-0593(2011)03-0816-04.
[14] 周瑛, 叶丽, 竹鑫平. HPLC-ICP-MS在食品中硒和砷形态分析及其生物有效性研究中的应用[J]. 化学进展, 2007, 19(6): 982-995.DOI:10.3321/j.issn:1005-281X.2007.06.014.
[15] WU P, BHATTARAI R R, DHITAL S, et al. In vitro, digestion of pectin- and mango-enriched diets using a dynamic rat stomachduodenum model[J]. Journal of Food Engineering, 2017, 202: 65-78.DOI:10.1016/j.jfoodeng.2017.01.011.
[16] SUN L, LIU G, YANG M, et al. Bioaccessibility of cadmium in fresh and cooked Agaricus blazei Murill assessed by in vitro, biomimetic digestion system[J]. Food & Chemical Toxicology, 2012, 50(5): 1729-1733. DOI:10.1016/j.fct.2012.02.044.
[17] LI S X, LIN L X, ZHENG F Y, et al. Metal bioavailability and risk assessment from edible brown alga Laminaria japonica, using biomimetic digestion and absorption system and determination by ICP-MS[J]. Journal of Agricultural & Food Chemistry, 2011, 59(3):822-828. DOI:10.1021/jf103480y.
[18] WAGH P, PARUNGAO G, VIOLA R E, et al. A new technique to fabricate high-performance biologically inspired membranes for water treatment[J]. Separation & Purification Technology, 2015, 156(2): 754-765. DOI:10.1016/j.seppur.2015.10.073.
[19] 林路秀, 李顺兴, 郑凤英. 应用体外仿生模型分析海藻水煎液中微量金属的形态和生物可给性[J]. 分析化学, 2010, 38(6): 823-827.DOI:10.3724/SP.J.1096.2010.00823.
[20] WÓJCIAK-KOSIOR M, SZWERC W, STRZEMSKI M, et al.Optimization of high-resolution continuum source graphite furnace atomic absorption spectrometry for direct analysis of selected trace elements in whole blood samples[J]. Talanta, 2017, 165: 351-356.DOI:10.1016/j.talanta.2016.12.077.
[21] ESKINA V V, DALNOVA O A, FILATOVA D G, et al. Separation and preconcentration of platinum-group metals from spent autocatalysts solutions using a hetero-polymeric S, N-containing sorbent and determination by high-resolution continuum source graphite furnace atomic absorption spectrometry[J]. Talanta, 2016, 159: 103-110.DOI:10.1016/j.talanta.2016.06.003.
[22] AJTONY Z, LACZAI N, DRAVECZ G, et al. Fast and direct screening of copper in micro-volumes of distilled alcoholic beverages by highresolution continuum source graphite furnace atomic absorption spectrometry[J]. Food Chemistry, 2016, 213: 799-805. DOI:10.1016/j.foodchem.2016.06.090.
[23] PERONICO V C D, RAPOSO J L. Ultrasound-assisted extraction for the determination of Cu, Mn, Ca, and Mg in alternative oilseed crops using flame atomic absorption spectrometry[J]. Food Chemistry, 2016,196: 1287-1292. DOI:10.1016/j.foodchem.2015.10.080.
[24] BECHLIN M A, FERREIRA E C, NETO J A G. Determination of chlorine in cement via CaCl molecule by high-resolution continuum source graphite furnace molecular absorption spectrometry with direct solid sample analysis[J]. Microchemical Journal, 2017, 132: 130-135.DOI:10.1016/j.microc.2017.01.019.
[25] 张建萍, 陈尚龙, 刘恩岐, 等. 微波消解-HR-CS AAS法测定几种调味品中的微量元素[J]. 现代食品科技, 2013, 29(6): 1424-1427.DOI:10.13982/j.mfst.1673-9078.2013.06.026.
[26] OOMEN A G, ROMPELBERG C J, BRUIL M A, et al. Development of an in vitro digestion model for estimating the bioaccessibility of soil contaminants[J]. Archives of Environmental Contamination &Toxicology, 2003, 44(3): 281-287. DOI:10.1007/s00244-002-1278-0.
[27] 陈安徽, 陈尚龙, 巫永华, 等. 黄精酵素口服液中钙、铁和锌的形态分析[J]. 现代食品科技, 2016, 32(1): 272-277. DOI:10.13982/j.mfst.1673-9078.2016.1.043.
[28] LI S X, ZHENG F Y, LIU X L, et al. Speciation analysis and the assessment of bioavailability of manganese in phytomedicines by extraction with octanol and determination by flame atomic absorption spectrometry[J]. Phytochemical Analysis Pca, 2005, 16(6): 405-410.DOI:10.1002/pca.858.
[29] 李勇, 陈尚龙, 王书兰, 等. 饮料酒中Cd的测定方法研究[J]. 徐州工程学院学报(自然科学版), 2012, 27(4): 16-19. DOI:10.15873/j.cnki.jxit.2012.04.005.
[30] EL HIMRI M, EL HIMRI A, PASTOR A, et al. Influence of the measurement medium and matrix modifiers on the determination of silicon in waters by graphite furnace atomic absorption spectrometry[J]. Journal of Applied Sciences and Environmental Management, 2013, 17(1): 37-41. DOI:10.4314/jasem.v17i1.
