导电碳修饰隔膜在高硫载量锂硫电池中的作用
2018-02-05裴海娟陈珠君解晶莹
裴海娟,郭 瑞,李 永,刘 雯,陈珠君,王 勇,解晶莹
导电碳修饰隔膜在高硫载量锂硫电池中的作用
裴海娟,郭 瑞,李 永,刘 雯,陈珠君,王 勇,解晶莹
(空间电源技术国家重点实验室,上海空间电源研究所,上海 200245)
研究了分别以乙炔黑、科琴黑、Super P、气相生长碳纤维、石墨烯、碳纳米管、XE-2为正极侧修饰层的隔膜对高硫载量(5.4 mg/cm2)锂硫电池电性能的影响。XE-2、Super P、石墨烯、科琴黑、气相生长碳纤维涂覆隔膜有助于提高隔膜正极侧的保液能力和导电能力;石墨烯、Super P涂层对多硫化锂有很好的阻挡、吸附作用。涂覆石墨烯的隔膜装配的电池的首次放电比容量可达1007 mA·h/g,远远高于采用空白隔膜的电池(340 mA·h/g),循环100次后放电比容量为539 mA·h/g,仍然高于采用空白隔膜的电池(399 mA·h/g)。
锂硫电池;隔膜;导电碳修饰;高硫载量
随着全球经济的持续增长,不可再生的化石燃料快速消耗,人类面临着资源和环境的双重危机。环境友好、可再生的新能源(风能、太阳能、潮汐能、地热能等)的利用离不开储能技术的发展。锂硫电池以其高能量密度、低成本、绿色环保的优势成为近年来倍受关注和投入较多研究的二次绿色化学电源[1-3]。锂硫电池的正极活性成分为单质硫、电解质为含锂盐的有机溶液、负极为金属锂。其电化学反应为:S8+Li=Li2S(1££8)=Li2S。按此反应计算,硫的理论比容量可达1675 mA·h/g,金属锂的理论比容量高达3860 mA·h/g,Li/S氧化还原对的理论能量密度高达2600 W·h/kg[4],可见二次锂硫电池在高比能电池方面具有相当诱人的应用前景。然而锂硫电池若想得到实际应用还有许多问题需要解决,如:正极活性材料导电性差、活性物质利用率低[5-7];放电中间产物多硫化锂易溶解,形成“穿梭效应”[8-10];金属锂负极在循环过程中快速消耗且易形成锂枝晶等[11]。
针对以上问题研究人员提出了很多解决方法:正极方面,将导电碳[12-16]、导电聚合物[8]、金属氧化 物[17-18]等以核壳结构或者多孔结构与硫材料复合来提高正极导电性、吸附溶解的多硫化锂、保持正极结构稳定;负极方面,采用金属锂表面修饰[19-20]、金属锂粉末化[21-22]、金属锂合金化[23-24]等方法来保护锂负极抑制锂枝晶的生成;电解液方面,通常采用加入功能型添加剂[25-26]、提高锂盐浓度[27-28]、采用固态电解 质[29,30]等技术手段来减缓多硫化锂溶解或促进金属锂表面形成保护膜。通过以上技术手段的应用,锂硫电池的正极活性物质利用率得到显著提高,多硫化锂的“穿梭效应”和锂枝晶的形成在一定程度上得到抑制,但仍不能满足实际需求。
隔膜作为电池的组成部分,其基本作用是隔离正、负极。锂硫电池特有的“穿梭效应”及锂枝晶问题,对锂硫电池隔膜提出了更高的要求。针对传统的聚烯烃类隔膜材料与电解液亲和性差、聚硫化锂阻挡能力弱的不足,研究人员采用对聚烯烃类隔膜进行改性的方法,在隔膜表面涂覆选择透过性材 料[31-32]、导电聚合物[33-34]、导电碳[35-36]、氧化物颗粒[37-38]等,阻挡、吸附多硫化锂[39]。选用导电碳材料作为隔膜涂层,除了可以阻挡多硫化物扩散至负极同金属锂发生反应之外,由于电池紧密装配、导电碳涂层同正极紧密接触,被导电碳材料吸附的多硫化锂可以继续反应,提高电池的放电比容量。本文主要研究了不同的商品碳材料作为隔膜修饰层对锂硫电池性能的影响。为了使锂硫电池高比能性能得到充分发挥,实际生产中对电极的硫载量提出很高要求,当电极硫载量提高后,活性材料的利用率往往会降低。本文以高硫载量正极为基础展开研究。
1 实 验
1.1 涂碳隔膜制备
本文所使用的导电碳材料为常用的商品碳材料,包括:乙炔黑(AB)、科琴黑EC600(KB)、Super P(SP)、气相生长碳纤维(VGCF)、石墨烯(G)、碳纳米管(CNT)、XE-2。
首先对两种黏结剂进行了筛选:PVDF和LA132。
以颗粒状SP和链状VGCF为例对黏结剂进行筛选。称取2.7 g LA132乳液(15%)和1.7 g去离子水用混料瓶在磁力搅拌器上搅拌至LA132完全溶解,分别称取0.05 g SP、VGCF、加入LA132溶液中,搅拌12 h后形成均一的黑色浆料。称取0.4 g PVDF 和4 g NMP用混料瓶在磁力搅拌器上搅拌至PVDF完全溶解,分别称取0.05 g SP、VGCF加入PVDF溶液中,搅拌12 h后形成均一的黑色浆料。
在自动涂膜机上将浆料涂布在Celgard2325隔膜上,将涂布后的隔膜放在55 ℃的鼓风烘箱里干 燥30 min。用17.5的冲子将涂碳隔膜冲成直径为17.5 mm的圆片,将隔膜转移至真空烘箱内真空干燥48 h。
1.2 涂碳隔膜性能表征
用四探针电阻测量仪测涂碳层的直流电阻;用测厚仪测含涂层的隔膜的厚度;用场发射扫描电子显微镜(HITACHI S-4800)对涂层形貌进行表征。
以PVDF为黏结剂,AB、KB、SP、VGCF、G、CNT、XE-2为导电剂(黏结剂与导电剂质量比为1∶1),按照上述方法制备涂碳隔膜并进行性能测试。
涂碳隔膜持液量水平测试:直径为17.5 mm的涂碳隔膜圆片的质量记为1,将其浸泡在电解液中1 小时后,取出称重,此时质量记为2,隔膜吸收的电解液的质量为2-1,隔膜持液率为(2-1)/1。
1.3 电池制备及电性能测试
正极极片主要由集流体和活性物质浆料涂层两部分组成,选铝箔为集流体。浆料由活性物质、黏结剂、导电剂和溶剂四部分构成。正极活性材料为化学法合成的碳/硫复合材料,活性材料含硫量为90%,黏结剂为LA132乳液,导电剂为SP(50%)和VGCF(50%),溶剂为去离子水。
正极极片的制备包括以下几个步骤:①混料:将材料按8∶1∶1的比例依次以黏结剂、导电剂、活性物质的顺序加入到去离子水中,搅拌均匀;②涂布:用自动涂膜机将混匀的浆料均匀地涂在铝箔上;③烘干:将已涂布好的铝箔置于鼓风烘箱中烘干后放入真空烘箱中烘干12 h;④辊轧:用塞尺调整辊轧机的紧度(100mm),将烘过的铝箔进行辊轧;⑤冲极片:用冲压模具将辊轧后的铝箔,冲成14 mm的极片,将冲好的极片放入真空烘箱烘干至少12h;⑥称重:用电子天平称20片14 mm的铝箔的总质量计算出每片铝箔的平均质量,称取每个极片的质量,用极片质量减去铝箔质量即可得到涂层质量。正极涂层单质硫面密度为5.4 mg/cm2。
扣式电池由电池壳、正极极片、负极极片、隔膜、电解液五部分组成。本实验中所用的负极极片为15.5 mm的金属锂片,隔膜有涂层的一侧对正极,电解液为1mol/L LiTFSI/DME∶DOL的体积比为1∶1,电池壳型号为CR2016。扣式电池的装配须在Ar气保护的手套箱内进行,装好之后在专用封口模具上封口成型。
在LAND CT2001A测试仪上进行充放电测试,电流密度为50 mA/g,充电截止电压为2.6 V,放电截止电压为1.5 V。
2 实验结果与讨论
2.1 黏结剂筛选
2.1.1 涂层形貌
图1为涂碳隔膜的SEM图。由图1可见,乙炔黑(AB)呈颗粒状、粒径约50 nm,堆积在隔膜表面;VGCF呈链状,直径约200 nm,分别对比图1(a)、图1(c),图1(b)、图1(d)可见,黏结剂LA132成块状,且占据较大面积;黏结剂PVDF则呈颗粒状。

图1 不同黏结剂/导电碳隔膜涂层的SEM图:(a)PVDF/AB;(b)PVDF/VGCF;(c)LA132/AB;(d)LA132/VGCF
2.1.2 涂层物理参数
表1记录了不同黏结剂/导电碳隔膜涂层的厚度、电阻值、透气度。其中透气度代表通过100 cm3空气所需时间。由表1可见涂碳层的导电性排序为:PVDF/VGCF>PVDF/AB>LA132/VGCF>LA132/AB;透气度排序为:PVDF/VGCF>PVDF/AB,而LA132/AB和LA132/VGCF则因透气性太差,无法测得透气度值。结合SEM图,块状的LA132黏结剂在隔膜表面的覆盖度远远高于颗粒状的PVDF黏结剂,因此对隔膜的透气性产生极大影响。隔膜透气性太差会影响电池的离子传输能力,使电池极化变大,放电电压平台下降,放电比容量降低,因此,我们选择PVDF作为隔膜涂层的黏结剂。

表1 涂层物理参数
2.2 不同碳材料隔膜涂层
2.2.1 涂层形貌表征
图2为不同碳材料涂碳隔膜的SEM图。由图2可见,石墨烯呈片状,层层叠叠平铺在隔膜表面;碳纳米管团聚成球状的二次颗粒;VGCF呈链状,直径约200 nm,被颗粒状的黏结剂黏成不均匀的 网状分布在隔膜表面;乙炔黑、科琴黑、Super P、XE-2碳均呈颗粒状、粒径约40 nm,堆积在隔膜 表面;科琴黑呈颗粒状,粒径约30 nm,堆积在隔膜表面。

(a)G

图2 不同碳材料隔膜涂层的SEM图
2.2.2 涂层物理参数
表2记录了不同碳材料涂层的质量、厚度、面密度、碳载量、透气度、方阻、持液率值。由表2可见,当涂层厚度为7mm时,AB、SP、XE-2、KB、CNT、VGCF、G涂层的面密度分别为0.127 g/μm、0.126 g/μm、0.111 g/μm、0.094 g/μm、0.144 g/μm、0.091 g/μm、0.041 g/μm,方阻值分别为581 Ω/sq、410 Ω/sq、595 Ω/sq、586 Ω/sq、284 Ω/sq、265 Ω/sq、51 Ω/sq,透气度分别为2016 s/100 cm3、3504 s/100cm3、786 s/100cm3、935 s/100cm3、1120 s/100cm3、3975 s/100cm3、978 s/100cm3,持液率分别为9.15%、11.02%、10.21%、7.18%、4%、7.22%、13.92%。当厚度相同时,涂层面密度:G

表2 不同涂碳隔膜的物理参数
2.3 电池电性能测试
图3~图4列出碳材料涂覆隔膜所制备的扣式电池的电性能测试结果。图3为空白隔膜以及不同碳材料(AB、G、CNT、KB、CNT、KB、SP、XE-2、VGCF)涂覆隔膜所制备的扣式电池的放电比容量-循环性能曲线。图4为空白隔膜以及不同碳材料涂覆隔膜所制备的扣式电池的首次放电曲线以及第2、20、40、60、100次充放电曲线。

图3 空白隔膜以及碳材料涂覆隔膜所制备的扣式电池放电比容量-循环次数曲线

图4 空白隔膜以及碳材料涂覆隔膜所制备的扣式电池不同循环次数的充放电曲线
由图3可见G涂覆隔膜扣式电池首次放电比容量即达到最大值为1007 mA·h/g,之后至第15次循环放电比容量迅速衰减,15~45次循环放电比容量稳定在600 mA·h/g左右,45~100次循环放电比容量稳定在550 mA·h/g左右,100次循环后放电比容量约为539 mA·h/g。空白隔膜以及AB、CNT、SP、CNT、KB、XE-2、VGCF涂覆隔膜电池首次放电时放电比容量都未达到最大值,随着循环次数的增加放电比容量逐渐增加达到最大值后又逐渐降低,只是达到最大值的循环次数有所不同。表3列出了不同隔膜所制备的电池达到最大放电比容量的循环次数及最高放电比容量,以及首次和循环100次时的放电比容量。理论上讲,由于放电中间产物多硫化锂的溶解作用,每次放电之后,多硫化锂都会有一部分溶解到电解液中扩散至负极,充电时无法回到正极生成单质硫,从而导致充、放电比容量随着循环的进行不断下降。下文结合电池的充放电曲线对本实验电性能结果作出详细分析。
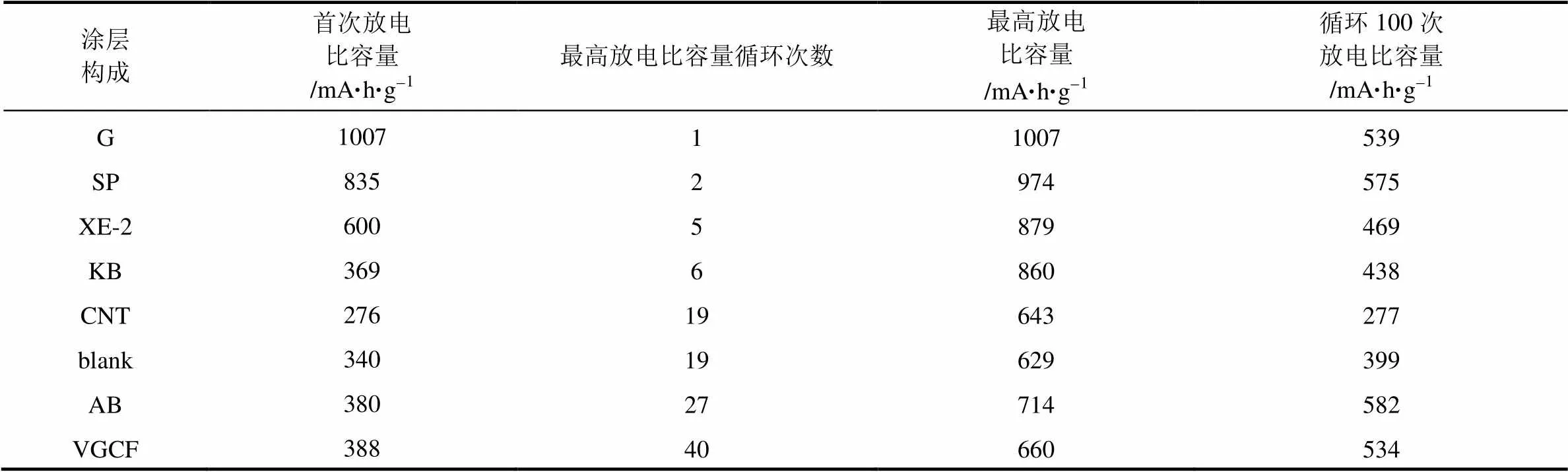
表3 空白隔膜以及碳材料涂覆隔膜所制备扣式电池放电 比容量-循环次数数据表
由图4可见除G涂覆隔膜外,空白隔膜及其它碳材料涂覆隔膜制备的电池的首次放电曲线第二个放电平台都较短,但随着循环次数增长第二个放电平台都逐渐增长。锂硫电池第一个放电平台对应的反应是单质硫同锂离子反应生成可溶的多硫化锂,其理论放电比容量为419 mA·h/g,第二个平台对应的反应为多硫化锂继续反应生成Li2S,其理论放电比容量为第一个平台对应的理论比能量的3倍,为1256 mA·h/g[40]。通过分析两个平台所对应的放电比容量,可以更详细地推断放电过程两个阶段的活性物质利用情况。
表4列出了空白隔膜及碳材料涂覆隔膜所制备的扣式电池分段比容量数据。第一平台所对应的放电比容量(1)反映了单质硫的反应情况,第一平台的理论放电比容量为419 mA·h/g,记为1*,1/1*即为单质硫的反应率。第二平台所对应的放电比容量(2)反映了可溶性多硫化锂的反应情况,由于第二个平台参与反应的多硫化锂来自第一个平台的反应产物,因此在讨论多硫化锂的反应率时应排除掉由于第一个平台单质硫未完全反应造成的对多硫化锂的损失,不能简单地以第一个平台完全反应时所对应的第二个平台的理论比能量计算,应该以第一个平台实际比能量来推算第二个平台具体的理论比能量。第二平台具体的理论比容量应为第一平台实际比容量的3倍,2*=1×3,2/2*即为多硫化锂的反应率。空白隔膜电池首次放电比容量为340 mA·h/g,其中1为230 mA·h/g,2为110 mA·h/g;最高放电比容量为629 mA·h/g,其中1为188 mA·h/g,2为441 mA·h/g;循环100次时放电比容量为399 mA·h/g,其中1为91 mA·h/g,2为308 mA·h/g。可见随着循环次数的增长,单质硫的利用率逐渐降低,而多硫化锂的利用率逐渐升高。这是因为随着循环次数的增加,单质硫不断消耗,1值降低;而电解液中的多硫化锂浓度不断提高,多硫化锂的溶解、扩散速度降低,多硫化锂的利用率(2/2*)提高。空白隔膜循环100次后2/2*大于1说明除了第一个放电平台生成的多硫化锂外电解液中溶解的多硫化锂也参与了反应。
1(首次放电)比1(最高放电比容量)高出42 mA·h/g,而2(首次放电)比2(最高放电比容量)低331 mA·h/g,这说明了造成空白隔膜电池首次放电比容量低的主要原因在于首次放电时多硫化锂的利用率过低。对比首次放电时不同涂碳隔膜的多硫化锂利用率发现,除G、SP涂覆隔膜外,其它隔膜电池的多硫化锂利用率都仅为20%左右,当放电比容量达到最大值时其多硫化锂利用率也都显著提升,达到了70%左右,与空白隔膜趋势一致。而G、SP涂覆隔膜首次放电时多硫化锂的利用率就达到了72%、53%,说明G、SP涂层对多硫化锂有很好的阻挡、吸附作用,且由于G、SP本身具有导电性,有助于多硫化锂进一步参与反应。如图4所示,G涂覆隔膜电池的首次放电曲线在第二个平台放电至650 mA·h/g时,放电电压出现回升,之后又持续放电至1007 mA·h/g,说明之前扩散至电解液中的多硫化锂被隔膜涂层吸附后又参与了反应。
由表4可见,首次放电过程中,对比1/1*可见单质硫的反应率:XE-2(0.81)>SP(0.77)> G(0.76)>KB(0.63)>VGCF(0.62)>blank(0.55)>AB(0.51)>CNT(0.39),空白隔膜单质硫反应率为55%,XE-2、SP、G、KB、VGCF涂覆隔膜电池单质硫反应率都有所提高。由于正极单质硫载量很高(5.4 mg/cm2),对电解液的需求量也很高,如果电解液不能够浸润极片就会导致单质硫无法充分反应,隔膜涂覆导电碳一方面有助于提高隔膜正极侧的保液能力,另一方面也有助于提高正极表面的导电能力,因此可以提高单质硫反应率。
综上所述,限制空白隔膜及大多数涂碳隔膜首次放电比容量发挥的原因有两个:① 首次放电时电解液对正极浸润程度不够,位于极片内部的单质硫无法完全反应;②位于极片表面参与反应的单质硫生成的多硫化锂极易溶解在电解液中、扩散至负极,难以继续反应,造成容量损失。XE-2、SP、G、KB、VGCF涂覆隔膜有助于提高隔膜正极侧的保液能力和导电能力,可以提高单质硫反应率。G、SP涂层对多硫化锂有很好的阻挡、吸附作用,且本身具有导电性,有助于多硫化锂进一步参与反应,因此可以提高多硫化锂利用率。

表4 空白隔膜以及碳材料涂覆隔膜所制备的扣式电池分段比容量数据表
表中为总放电比容量,1、2分别为放电曲线第一、第二平台所对应比容量,1*、2*分别为放电曲线第一、第二平台所对应理论比容量,、1、2、1*、2*单位为mA·h/g
3 结 论
(1)高硫载量含硫正极通常涂层较厚,首次放电时电解液对正极浸润程度不够,极片内部单质硫无法完全反应,而位于极片外侧参与反应的单质硫生成的放电中间产物多硫化锂更容易溶解在电解液中,扩散至负极,造成容量严重损失。
(2)XE-2、SP、G、KB、VGCF涂覆隔膜有助于提高隔膜正极侧的保液能力和导电能力,可以提高单质硫反应率。
(3)G、SP涂层对多硫化锂有很好的阻挡、吸附作用,且本身具有导电性,有助于多硫化锂进一步参与反应,可以提高多硫化锂利用率。
(4)隔膜表面涂覆G可以使正极的首次放电比容量提升约2倍,循环100此后放电比容量提升35%;隔膜表面涂覆SP可以使正极的首次放电比容量提升约1.5倍,循环100此后放电比容量提升44%。
[1] BRUCE P G, FREUNBERGER S A, HARDWICK, et al. Li-O2and Li-S batteries with high energy storage[J]. Nature Materials. 2011, 11: 19-29.
[2] YANG Y, ZHENG G, CUI Y. Nanostructured sulfur cathodes[J]. Chemical Society Reviews, 2013, 42: 3018-3032.
[3] MANTHIRAM A, FU Y, CHUNG S H, et al. Rechargeable lithium-sulfur batteries[J]. Chemical Reviews, 2014, 114: 11751-11787.
[4] GAO X P, YANG H X. Multi-electron reaction materials for high energy density batteries[J]. Energy & Environmental Science. 2010, 3: 174-189.
[5] SUN Z J, XIAO M, WANG S J, et al. Specially designed carbon black nanoparticle-sulfur composite cathode materials with a novel structure for lithiumesulfur battery application[J]. Journal of Power Sources 2015, 285: 478-484.
[6] YIN Y X, XIN S, GUO Y G, et al. Lithium-sulfur batteries: Electrochemistry, materials, and prospects[J]. Angewandte Chemie International Edition, 2013, 52: 13186-13200.
[7] JI X L, LEE K T, NAZAR L F. A highly ordered nanostructured carbon sulphur cathode for lithium-sulphur batteries[J]. Nature Materials, 2009, 8: 500-506.
[8] LI G C, LI G R, YE S H, et al. A polyaniline-coated sulfur/carbon composite with an enhanced high-rate capability as a cathode material for lithium/sulfur batteries[J]. Advanced Energy Materials, 2012, 2: 1238-1245.
[9] ZHANG S G, UENO K, DOKKO K, et al. Recent advances in electrolytes for lithium-sulfur batteries[J]. Advanced Energy Materials, 2015, 5:doi: 10.1002/aenm.201500117.
[10] JI X, EVERS S, BLACK R, et al. Stabilizing lithium-sulphur cathodes using polysulphide reservoirs[J]. Nature Communications, 2011, 2: doi:10.1038/ncomms1293.
[11] CHENG X B, ZHANG R, ZHAO, C Z, et al. A review of solid electrolyte interphases on lithium metal anode[J]. Advanced Science, 2016, 3: doi: 10.1002/advs.201500213.
[12] SHI J L, TANG C, PENG H J, et al. 3D mesoporous graphene: CVD self-assembly on porous oxide templates and applications in high-stable Li-S batteries[J]. Small, 2015, 11: 5243-5252.
[13] SONG J X, GORDIN M L, XU T, et al. Strong lithium polysulfide chemisorption on electroactive sites of nitrogen-doped carbon composites for high-performance lithium-sulfur battery cathodes[J]. Angewandte Chemie International Edition, 2015, 54: 4325-4329.
[14] PENG H J, WANG D W, HUANG J Q, et al. Janus structures: Janus separator of polypropylene-supported cellular graphene framework for sulfur cathodes with high utilization in lithium-sulfur batteries[J]. Advanced Science, 2016, 3: doi:10.1002/advs.201500268.
[15] OH Y S, JUNG G Y, KIM J H. Janus-faced, dual-conductive/ chemically active battery separator membranes[J]. Advanced Functional Materials, 2016, 26(39): 7074-7083.
[16] LUO L, CHUNG S H, MANTHIRAM A. A trifunctional multi-walled carbon nanotubes/polyethylene glycol (MWCNT/PEG)- coated separator through a layer-by-layer coating strategy for high-energy Li-S batteries[J]. Journal of Materials Chemistry A, 2016, 4: 16805-16811.
[17] LI Y, YE D X, LIU W, et al. A MnO2/graphene oxide/multi-walled carbon nanotubes-sulfur composite with dual-efficient polysulfide adsorption for improving lithium-sulfur batteries[J]. ACS Applied Materials & Interfaces, 2016, 8: 28566-28573.
[18] VIDA L, NATASA N T, ALENKA R, et al. Manganese modified zeolite silicalite-1 as polysulphide sorbent in lithium sulphur batteries[J]. Journal of Power Sources, 2015, 274: 1239-1248.
[19] ZHENG G, LEE S W, LIANG Z, et al. Interconnected hollow carbon nanospheres for stable lithium metal anodes[J]. Nature Nanotechnology, 2014, 9(8): 618-623.
[20] YAN K, LEE H W, GAO T, et al. Ultrathin two-dimensional atomic crystals as stable interfacial layer for improvement of lithium metal anode[J]. Plant & Cell Physiology, 2009, 14(10): 2210-2222.
[21] KWON C W, CHEON S E, SONG J M, et al. Characteristics of a lithium-polymer battery based on a lithium powder anode[J]. Journal of Power Sources, 2001, 93(1): 145-150.
[22] JIN S K, YOON W Y. Improvement in lithium cycling efficiency by using lithium powder anode[J]. Electrochimica Acta, 2004, 50(2/3): 531-534.
[23] ISHIKAWA M, YOSHITAKE S, MORITA M, et al. In situ scanning vibrating electrode technique for the characterization of interface between lithium electrode and electrolytes containing additives[J]. Journal of the Electrochemical Society, 1994, 141(12): L159-L161.
[24] CHEN C, YANG Y, SHAO H. Enhancement of the lithium cycling capability using Li-Zn alloy substrate for lithium metal batteries[J]. Electrochimica Acta, 2014, 137(8): 476-483.
[25] XIONG S, DIAO Y, HONG X, et al. Characterization of solid electrolyte interphase on lithium electrodes cycled in ether-based electrolytes for lithium batteries[J]. Journal of Electroanalytical Chemistry, 2014, 719(4): 122-126.
[26] QIAN J, XU W, BHATTACHARYA P, et al. Dendrite-free Li deposition using trace-amounts of water as an electrolyte additive[J]. Nano Energy, 2015, 15: 135-144.
[27] AURBACH D, ZABAN A. Impedance spectroscopy of lithium electrodes: Part 1. General behavior in propylene carbonate solutions and the correlation to surface chemistry and cycling efficiency[J]. Journal of Electroanalytical Chemistry, 1993, 348(1/2): 155-179.
[28] SUO L, HU Y S, LI H, et al. A new class of solvent-in-salt electrolyte for high-energy rechargeable metallic lithium batteries[J]. Nature Communications, 2013, 4(2): 66-78.
[29] SCROSATI B, SELYAGGI A, CROCE F, et al. The Li/LiV3O8polymer electrolyte lithium battery III. Investigation of the electrode interfaces[J]. Journal of Power Sources, 1988, 24(4): 287-294.
[30] AGRAWAL R C, PANDEY G P. Solid polymer electrolytes: Materials designing and all-solid-state battery applications: an overview[J]. Journal of Physics D: Applied Physics, 2008, 41(22): 3715-3725.
[31] BAUER I, THIEME S, BRUCKNER J, et al. Reduced polysulfide shuttle in lithium-sulfur batteries using Nafion-based separators[J]. Journal of Power Sources, 2014, 251: 417-422.
[32] GU M, LEE J Y, KIM Y, et al. Inhibiting the shuttle effect in lithium-sulfur batteries using a layer-by-layer assembled ion-permselective separator[J]. RSC Advances, 2014, 4(87): 46940- 46946.
[33] CHANG C H, CHUNG S H, MANTHIRAM A. Ultra-lightweight PANiNF/MWCNT-functionalized separators with synergistic suppression of polysulfide migration for Li-S batteries with pure sulfur cathodes[J]. Journal of Materials Chemistry A, 2015, 3: 18829-18834.
[34] NATSUKI N , TOKIHIKO Y , HIROKI N, et al. Suppression of polysulfide dissolution by polypyrrole modification of sulfur-based cathodes in lithium secondary batteries[J]. Journal of Power Sources, 2015, 74: 1263-1266.
[35] LI W, CHEN Y F, LI P J, et al. Enhanced performance of lithium sulfur battery with a reduced graphene oxide coating separator journal of the electrochemical society[J]. Journa1 of the Electrochemical Society, 2015, 62 (8): A1624-A1629 .
[36] ZHANG Z Y, LAI Y Q, ZHANG Z A, et al. A functional carbon layer-coated separator for high performance lithium sulfur batteries[J]. Solid State Ionics, 2015, 278: 166-171.
[37] LIU X Y, SHAN Z Q, ZHU K L, et al. Sulfur electrode modified by bifunctional nafion/γ-Al2O3membrane for high performance lithium-sulfur batteries[J]. Journal of Power Sources, 2015, 274: 85-93.
[38] XU Q, HU G C, BI H L, et al. A trilayer carbon nanotube/Al2O3/ polypropylene separator for lithium-sulfur batteries[J]. Ionics, 2015, 21(4): 981-986.
[39] 许睿, 赵梦, 黄佳琦. 复合隔膜在锂硫电池中的应用评述[J]. 储能科学与技术, 2017, 6(3): 433-450.
XU R, ZHAO M, HUANG J Q. Flexible cathodes for lithium sulfur battery: A review[J]. Energy Storage Science and Technology, 2017, 6(3): 433-450.
[40] CE L B, FLORIAN M, CAROLE D, et al. Lithium/sulfur cell discharge mechanism: An original approach for intermediate species identification[J]. Analytical Chemistry, 2012, 84: 3973-3980.
Conductive carbon-coated separator for high sulfur-loading lithium sulfur batteries
PEI Haijuan, GUO Rui, LI Yong, LIU Wen, CHEN Zhujun, WANG Yong, XIE Jingying
(State Key Laboratory of Space Power Technology, Shanghai Institute of Space Power-sources, Shanghai 200245, China)
The effects of separators consisting of cathode-side functional layers separately composed of acetylene black, Ketjen black, Super P, vapor-grown carbon fiber, graphene, carbon nano-tube and XE-2, on high sulfur-loading(5.4 mg/cm2)lithium sulfur batteries have been investigated. The separators coated by XE-2, Super P, graphene, ketjen black, vapor-grown carbon fiber can improve the electrolyte adsorbility and electrical conductivity of cathode side. Besides, the separators coated by Super P and graphene can obstruct and adsorb lithium polysulfide. The battery with separator coated by graphene delivers a specific capacity as high as 1007 mA·h/g, much higher than that of untreated separator (340 mA·h/g). After 100 cycles, the battery with separator coated by graphene can still maintain a specific discharge capacity of 539 mA·h/g, still higher than that of untreated separator (399 mA·h/g).
lithium-sulfur battery; separator; conductive carbon-coated; high sulfur-loading
TM 912.9
A
2095-4239(2018)01-056-10
10.12028/j.issn.2095-4239.2017.0053
2017-05-08;
2017-06-08。
国家自然科学基金项目(21373137)。
裴海娟(1986—),女,工程师,主要研究方向为锂硫电池正极材料、隔膜材料,E-mail:phjgwt613@126.com;
解晶莹,研究员,主要研究方向为锂离子电池、新型动力电池、金属锂电池等,E-mail:xiejingying2007@126.com。
