酰胺-亚胺醇互变诱导的分子内1,3-羟基迁移裂解反应研究
2018-01-22许杨洁曹小吉
许杨洁,曹小吉,2*
(1.浙江工业大学 化学工程学院,杭州310014; 2.浙江工业大学 分析测试中心,杭州310014)
酰胺键是构成多肽和蛋白质的基本骨架结构[1]。在25%的药物分子中都含有酰胺键片段。酰胺-亚胺醇互变类似于酮-烯醇互变,区别在于其C=N双键代替了烯醇式中的C=C双键。尽管酰胺的亚胺醇构型在通常情况下是不稳定的,但是其作为一类特殊的反应中间体日益得到人们的关注。文献[2]报道采用量子化学理论计算证实了天冬氨酸残基的C-端肽键在两个水分子辅助下可以发生亚胺醇异构化,进而触发了天冬氨酸残基向其β构型转化。文献[3]报道采用红外多光子解离谱研究了二价金属离子诱导的酰胺-亚胺醇异构化反应,结果发现,在气相中金属离子可与亚胺醇上的氮原子形成稳定的配位键。此外,研究还发现O-(3,5-二硝基苯甲酰)苯甲羟肟酸酯与二甲亚砜共结晶,其酰胺键以亚胺醇式存在[4]。
N-吡啶基苯甲酰胺类化合物具有抗菌、抗肿瘤等生物活性[5]。截至目前,其质子化离子碰撞诱导解离产生取代2-羟基吡啶阳离子的反应机理研究还未见报道。碰撞诱导解离(CID)的原理是让具有一定动能的母离子与质谱仪中的惰性气体发生碰撞,动能转化为内能,当内能超过了母离子的解离能时,会发生碎裂。高能碰撞解离(HCD)技术是一种新型的质谱裂解技术,与CID技术相比,HCD技术具有稳定的高能量裂解方式,可产生更多的碎片离子以及无低质量端歧视效应等优点,与静电场轨道阱质量分析器结合使用还可以获得高分辨质谱数据,确定碎片离子的元素组成[6-7]。
本工作采用高分辨的电喷雾电离-高能碰撞解离质谱(ESI-HCD-MS/MS)技术,结合同位素标记试验和量子化学密度泛函理论计算,系统探讨了酰胺-亚胺醇互变诱导的分子内羟基迁移解离产生取代2-羟基吡啶阳离子的反应机理。
1 试验部分
1.1 仪器与试剂
Thermo Fisher LCQ Deca XP MAX离子阱质谱仪,配电喷雾电离源(ESI)及Xcalibur 2.1数据处理系统;Thermo Fisher Q Exactive组合型四极杆Orbitrap质谱仪(分辨率大于7 000,FWHM),配H-ESI II源及 Xcalibur 2.2数据处理系统。
N-(吡啶-2-基)苯甲酰胺、N-(6-溴吡 啶-2-基)苯甲酰胺、N-(6-氯吡啶-2-基)苯甲酰胺、4-甲基-N-(吡啶-2-基)苯甲酰胺、4-溴-N-(吡啶-2-基)苯甲酰胺、4-甲氧基-N-(吡啶-2-基)苯甲酰胺、4-甲基-N-(6-甲基吡啶-2-基)苯甲酰胺等7个N-(吡啶-2-基)苯甲酰胺类化合物纯度均大于95%。
甲醇为色谱纯。
1.2 仪器工作条件
1)LCQ Deca XP MAX离子阱质谱条件 电喷雾正离子(ESI+)扫描模式,扫描范围m/z50~800,流量5μL·min-1;雾化气为氮气,流量3L·min-1;喷雾电压4.0kV;金属毛细管温度300℃;碰撞气为氦气,碰撞能量为32%。
2)Q Exactive组合型四极杆Orbitrap质谱条件 正离子模式,流量为3μL·min-1;雾化气为氮气,流量3L·min-1;喷雾电压3.0kV;金属毛细管温度240℃;碰撞气为氮气,碰撞能量为35%;采用标 准 校 正 液 (caffenine,MRFA and Ultramark 1621)对质量轴进行校正。
1.3 试验方法
1.3.1 样品制备
称取样品2~3μg溶于1mL甲醇中,过0.45μm滤膜,用注射泵输送至质谱仪分析。
1.3.2 计算条件
采用Gaussian 03软件,密度泛函理论(DFT),在B3LYP/6-31G(d)水平上对分子结构进行模拟和优化。在构型优化过程中,忽略对称性限制。对所有的优化结构进行振动频率分析,其中过渡态有且仅有一个虚频。计算所得的能量为电子能和内能之和。过渡态结构优化完成后通过反应内禀坐标(IRC)进行验证。
2 结果与讨论
2.1 N-(吡啶-2-基)苯甲酰胺的ESI-MS/MS分析
7个N-(吡啶-2-基)苯甲酰胺类化合物的化学结构式见表1。
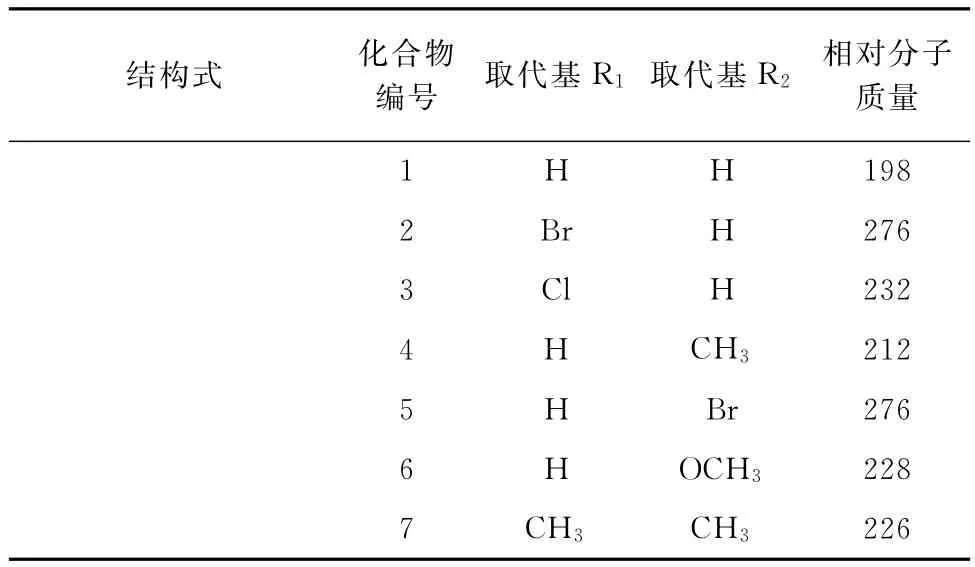
表1 N-吡啶基苯甲酰胺类化合物的结构式Tab.1 Structural formulae of the N-pyridinyl benzamides
在ESI+下,表1中的化合物均产生质子化离子[M+H]+,其质子化离子碰撞诱导解离(CID-MS/MS)裂解所产生的主要碎片离子见表2。
由表2可知:这类化合物的质子化离子碰撞诱导解离,裂解模式相同。
化合物1~7的质子化离子及碎片离子的高分辨质谱数据见表3。
由表3可知:高分辨ESI-HCD-MS/MS得到的碎片离子的精确质量数的误差均小于4×10-6,确证主要生成了取代2-羟基吡啶阳离子a、取代苯甲酰阳离子b以及中性丢失一分子水产生的碎片离子c,它们的丰度随着取代基的变化而变化。
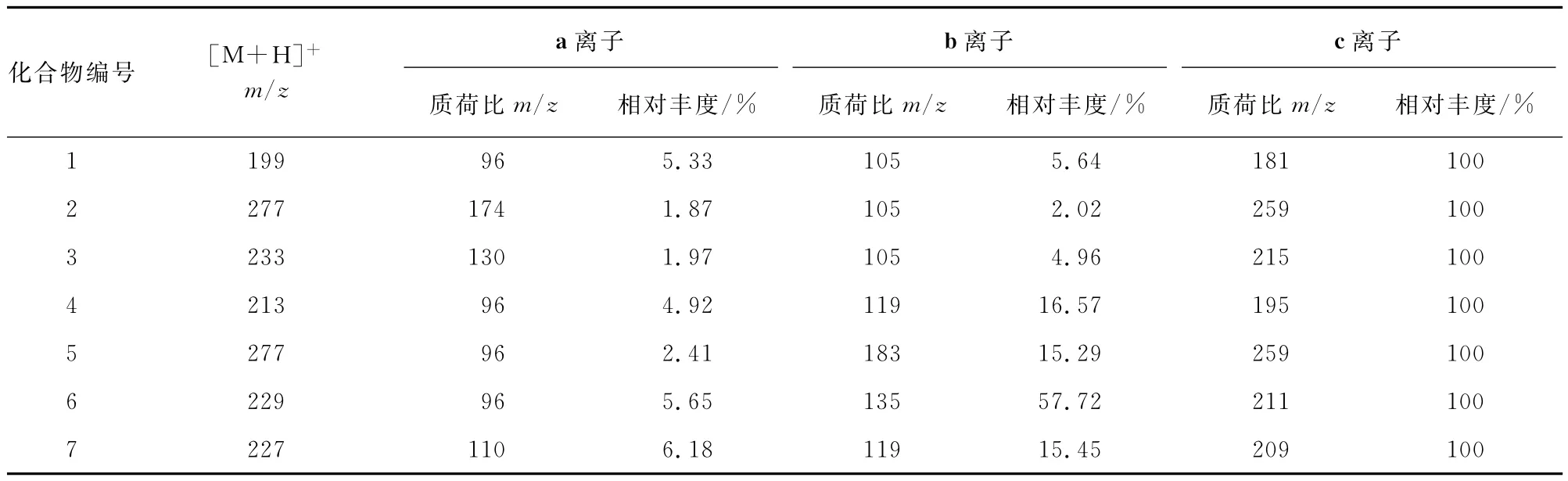
表2 N-吡啶基苯甲酰胺类化合物质子化离子CID-MS/MS产生的主要碎片离子数据Tab.2 CID-MS/MS data of major fragement ions from protonated N-pyridinyl benzamides
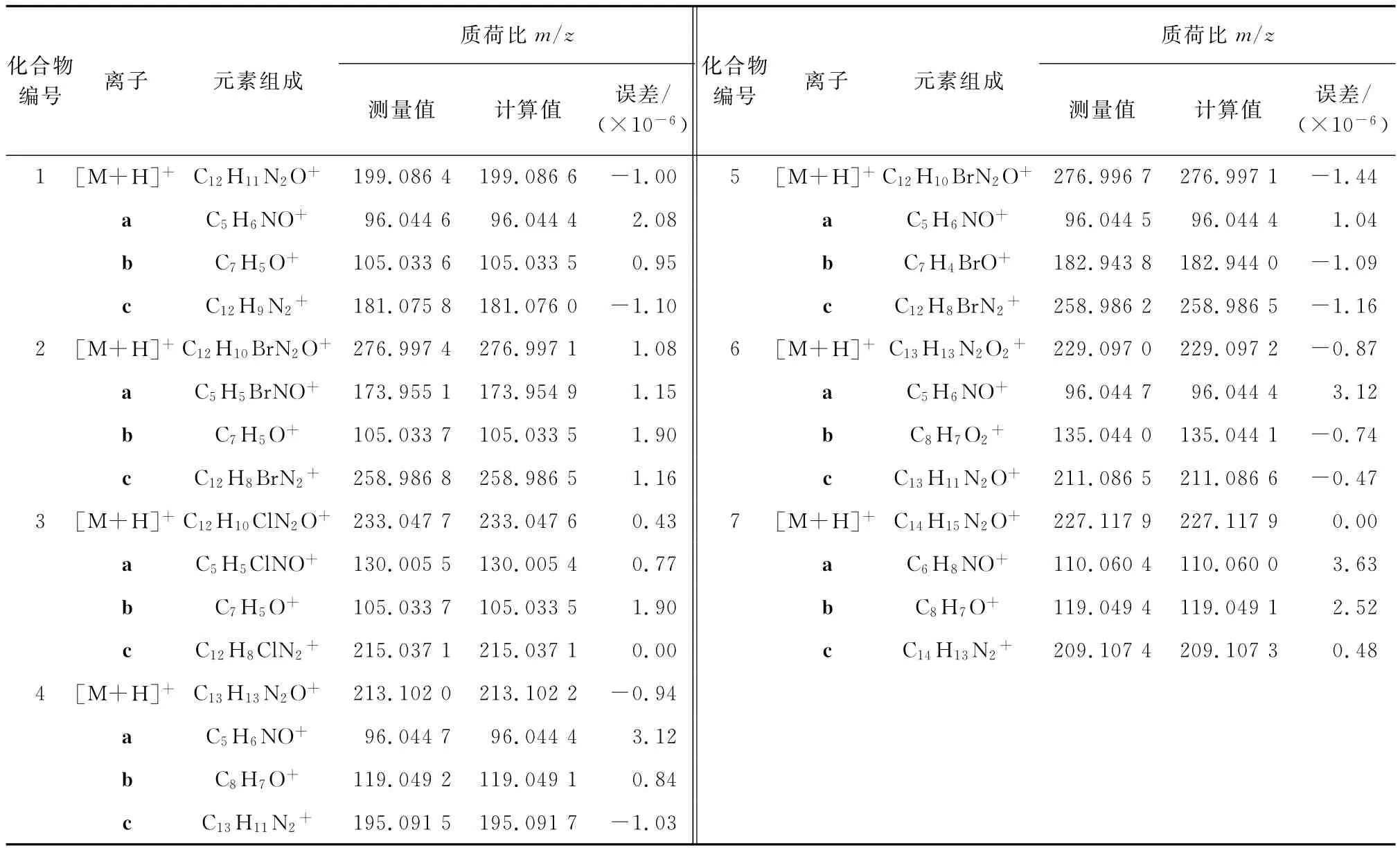
表3 化合物1~7的高分辨HCD-MS/MS数据Tab.3 High-resolution HCD-MS/MS data of compounds 1-7
2.2 酰胺-亚胺醇互变诱导的1,3-OH-迁移裂解反应
试验以化合物7为例探讨碎片离子a的具体形成机理。
化合物7的高能碰撞解离二级质谱(HCD-MS/MS)图见图1(a),化合物7的[M+H]+离子(m/z227.117 9)碰撞解离产生6-甲基-2-羟基-吡啶阳离子am/z110.060 4(元素组成C6H8NO+,理论m/z110.060 0,相对误差3.63×10-6);活泼氢完全氘代的化合物7的[M-d1+D]+离子(m/z229.2)碰撞诱导解离则产生活泼氢完全氘代的a-d2碎片离子(m/z112.2)见图1(b),表明酰胺上的活泼氢参与了6-甲基-2-羟基吡啶阳离子的生成;为了推导碎片离子a中氧原子的来源,试验还研究分析了18O标记的化合物1的质子化离子(m/z201.090 8)的HCD-MS/MS图,图1(c)中m/z98.048 5的信号(元素组成C5H6N18O+,相对误差为-1.04×10-6)为18O标记的2-羟基吡啶阳离子,说明2-羟基吡啶阳离子中的氧原子来自于苯甲酰基上的酰基氧。以上数据表明:化合物7的质子化离子必须通过分子内的骨架重排反应,将酰胺键上的氧原子和活泼氢原子迁移到吡啶环上才能裂解产生碎片离子a。
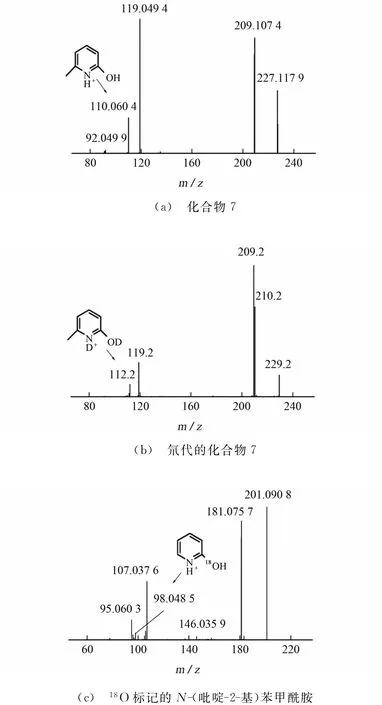
图1 ESI-MS/MS质谱图Fig.1 ESI-MS/MS spectra
酰胺键的酰胺-亚胺醇互变与酮的酮-烯醇互变类似,其亚胺醇式比酰胺式不稳定。试验和量子化学理论计算已经证实在气相肽键的互变异构化过程中存在亚胺醇构型[8-9]。由量子化学密度泛函理论计算可知,当外加质子加合到化合物7的N1原子上时,吡啶环上的N1原子和酰胺键上的氧原子通过氢键螯合,产生质子化离子MH1,其能量比加合在C2、N3、C4、C5原子上产生的质子化离子 MH2、MH3、MH4、MH5的能量分别低89.58,139.96,208.22,203.82kJ·mol-1,由此可以确定吡啶环上的N1原子是该化合物热力学上最稳定的质子化位点。MH1(0kJ·mol-1)经碰撞活化,外加质子诱导,发生酰胺-亚胺醇异构化反应,产生亚胺醇式中间体 M(56.21kJ·mol-1),亚胺醇上的羟基经过过渡态 TS(377.85kJ·mol-1),发生1,3-OH-迁移重排到吡啶环的C2位上,正电荷诱导C2-N3键异裂,中性丢失一分子4-甲基苯腈(e)产生6-甲基-2-羟基吡啶阳离子(a),推测的具体裂解途径见图2,势能图见图3。
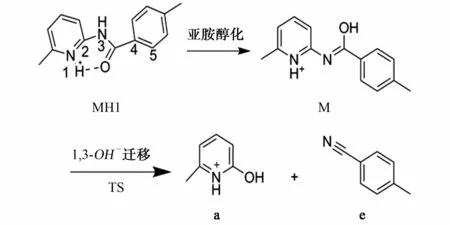
图2 推导的化合物7质子化离子产生离子a的裂解机理Fig.2 Proposed fragmentation mechanism of protonated compound 7to produce ion a
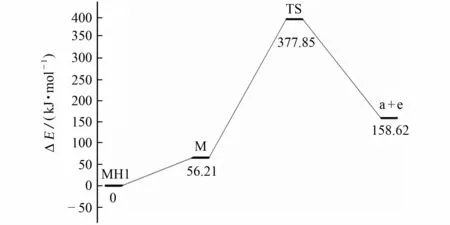
图3 化合物7生成离子a的反应势能图Fig.3 Potential energy diagram for fragmentation of compound 7to produce ion a
为了更准确地考察整个裂解反应的路径,试验对化合物1~7进行量子化学密度泛函理论计算,数据见表4。
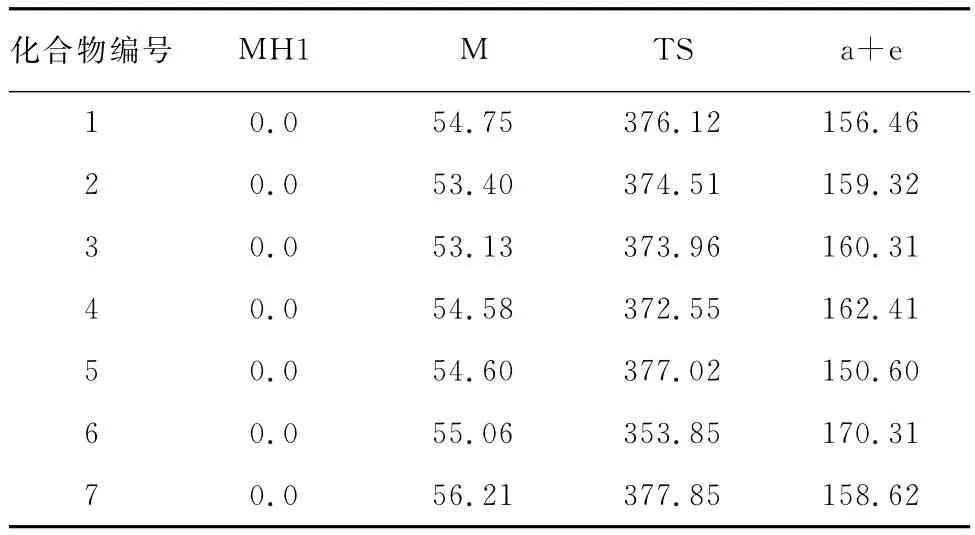
表4 化合物1~7的各阶段相对势能量Tab.4 Relative potential energies at various stages of compounds 1-7 kJ·mol-1
此外,试验还研究了取代基对该裂解反应的影响。结果表明:当吡啶环上有吸电子取代基或者苯环上有推电子取代基时,不利于该裂解反应发生,产物离子a的丰度减小。
本工作采用高分辨电喷雾串联质谱技术,结合同位素标记试验以及量子化学密度泛函理论计算,探讨了N-(吡啶-2-基)苯甲酰胺类化合物的质子化离子解离产生取代2-羟基吡啶阳离子的反应机理。研究发现,外加质子首先加合在吡啶环的氮原子上,随后诱导酰胺键发生酰胺-亚胺醇异构化转变为亚胺醇式,接着发生分子内的1,3-OH-重排,正电荷诱导异裂产生取代2-羟基吡啶阳离子。吡啶环上有吸电子取代基或苯环上有推电子取代基不利于该裂解反应的发生。本工作的研究结果对多肽以及蛋白质中肽键的酰胺-亚胺醇互变异构化机理的系统深入研究具有一定的理论指导意义。
感谢万结平副教授提供研究样品!
[1] 柴云峰.大气压电离质谱中含氮有机化合物质子化离子的产生及裂解反应研究[D].杭州:浙江大学,2013.
[2] OHGI T,AKIFUMI O.Tyrosine and aspartic acid[M].New York:Nova Science Publishers,Inc,2012.
[3] DUNBAR R C,STEILL J D,POLFER N C,et al.Peptide bond tautomerization induced by divalent metal ions:Characterization of the iminol configuration[J].Angewandte Chemie International Edition,2012,124(19):4669-4671.
[4] ALBRIGHT A L,COLLINS L,LI J,et al.Crystal structures of the amide and iminol tautomers o-(3,5-dinitrobenzoyloxy)benzohydroxamate.A case of a disappearing solvate?[J].Australian Journal of Chemistry,2016,69(10):1193-1197.
[5] PREMKUMAR S,REKHA T N,ASATH R M,et al.Vibrational spectroscopic,structural and quantum chemical studies on N-phenyl-3-pyridinecarboxamide[J].Journal of Molecular Structure,2016,1107:254-265.
[6] CHI H,SUN R X,YANG B,et al.pNovo:de novo peptide sequencing and identification using HCD spectra[J].Journal of Proteome Research,2010,9(5):2713-2724.
[7] ZUZANA S,JOHN O,FRANK N,et al.Food contaminant analysis at ultra-high mass resolution:Application of hybrid linear ion trap-orbitrap mass spectrometry for the determination of the polyether toxins,azaspiracids,in shellfish[J].Rapid Communications in Mass Spectrometry,2010,24(20):2966-2974.
[8] RACZYNSKA E D,HALLMANN M,DUCZMAL K.Quantum-chemical studies of amide-iminol tautomerism for inhibitor of lactate dehydrogenase:Oxamic acid[J].Computational and Theoretical Chemistry,2011,964(1/2/3):310-317.
[9] FAIRLIE D P,TAI C W,WICKRAMASINGHE W A,et al.Amide-iminol tautomerism:Effect of metalation[J].Inorganic Chemistry,1994,33(26):6425-6428.
