超高效液相色谱-串联四极杆飞行时间质谱法测定人体尿液中14种内源性甾体激素含量
2018-01-22潘虹
潘 虹
(齐齐哈尔医学院 药学院,齐齐哈尔161006)
甾体激素,又称类固醇激素,是一类具有环戊烷多氢菲母核的四环脂肪烃化合物,对机体生长、免疫调节、性器官发育和生殖控制等方面起关键作用[1-2]。外源性甾体激素是体外合成物质,如丙酸睾酮、苯甲酸雌二醇和乙酸美仑孕酮等,对机体体能恢复、肌肉增长、神经兴奋状态有重要作用,被国际反兴奋剂组织和国际奥林匹克委员会列为运动员禁用药物[3];内源性甾体激素是由体内内腺生成的激素,如睾酮、表睾酮、雄酮、脱氢表雄酮、孕酮、孕二醇、β-雌二醇等[4],在体内作为化学信使。随着国际竞技体育对兴奋剂的禁用,部分运动员选择使用隐蔽性更强的内源性甾体激素兴奋剂。因此,建立可靠有效的检测方法以监控内源性甾体激素在人体尿液中残留尤为必要。
目前关于甾体激素的测定方法主要有酶联免疫吸附法[1-2]、液相色谱法[5]、气相色谱-质谱法[6-7]、液相色谱-质谱法[8-10]等,其中酶联免疫吸附法和液相色谱法灵敏度低、选择性和特异性差;气相色谱-质谱法前处理需要衍生化,操作繁琐、耗时长;而液相色谱-质谱法无需衍生化,且灵敏度、选择性和特异性较高,已成为测定甾体激素残留量的重要方法。本工作使用乙醚超声萃取,经Oasis HLB固相萃取柱净化后,采用超高效液相色谱-串联四极杆飞行时间质谱法(UPLC-QToF/MS)测定人体尿液中雌酮、17α-雌二醇、17β-雌二醇、雌三醇、丙酸睾酮、雄酮、本胆烷醇酮、表睾酮、脱氢表雄酮、睾酮、孕酮、孕二醇、孕三醇、氢化可的松等14种内源性甾体激素残留量,可为行政部门监管内源性甾体激素兴奋剂滥用的判定提供检测依据。
1 试验部分
1.1 仪器与试剂
Waters Xevo G2-QToF型超高效液相色谱-串联四极杆飞行时间质谱仪(UPLC-QToF/MS);MS型分析天平(精度0.1mg);Milli-Q型超纯水器;JP-020S型超声波清洗仪;3-18K型冷冻离心机;Waters-Oasis HLB固相萃取柱;TurboVap LV 型氮吹仪;0.22μm聚四氟乙烯膜针头式过滤器。
雌酮、17α-雌二醇、17β-雌二醇、雌三醇、孕酮、孕三醇、丙酸睾酮、雄酮、本胆烷醇酮、脱氢表雄酮、睾酮、氢化可的松等标准品纯度均不小于99.5%;孕二醇、表睾酮的纯度均不小于98.0%;β-葡萄糖醛酸甙酶,5 000IU。
甾体激素标准品储备溶液:1 000mg·L-1,分别称取10.0mg甾体激素标准品,用甲醇溶解并定容至10mL,于-20℃以下保存。
磷酸盐缓冲溶液:称取十二水合磷酸氢二钠(Na2HPO4·12H2O)35.8g,二水合磷酸二氢钠(NaH2PO4·2H2O)15.6g,用水溶解并定容至1L,用0.01mol·L-1氢氧化钠溶液调节酸度为pH 6.8。
尿样品由某医院泌尿外科及志愿者提供;甲醇、乙腈、甲酸、乙酸铵均为色谱纯,试验用水为超纯水。
1.2 仪器工作条件
1)色谱条件 ACQUITY UPLC?HSS T3色谱柱(2.1mm×100mm,1.8μm),柱温45℃;进样量2μL;流量0.2mL·min-1;流动相A为含0.1%(体积分数,下同)甲酸的5mmol·L-1乙酸铵溶液,B为乙腈。梯度洗脱程序:0~6min时,B由95%降至90%;6~16min时,B由90%降至85%;16~20min时,B由85%降至60%。
2)质谱条件 Xevo G2-QtoF质谱系统,电喷雾离子源(ESI),正离子或负离子扫描模式,离子源温度120℃;毛细管电压2 500V;锥孔电压35V,锥孔气流量60L·h-1;雾化器温度500℃,雾化气流量800L·h-1;质量扫描范围m/z为50~500;全信息串联质谱(MSE)采集模式。其他质谱参数见表1。

表1 质谱参数Tab.1 MS parameters

表1(续)
1.3 试验方法
称取试样5.000 0g于50mL离心管中,加入pH 6.8的磷酸缓冲溶液2.0mL、β-葡萄糖醛酸甙酶150μL混合,于55℃恒温水浴中培养2h,取出后冷却至室温,再加入乙醚5mL,超声萃取5min,以10 000r·min-1转速离心10min,取出有机相;水相中再加入乙醚5mL,超声萃取5min,以10 000r·min-1转速离心10min,合并有机相,于45℃氮气吹干,加入甲醇(3+7)溶液5mL溶解残渣,用Oasis HLB固相萃取(SPE)柱净化,用甲醇-乙腈(1+1)溶液5mL洗脱,于45℃氮气吹干后,用乙腈(95+5)溶液溶解并定容至1mL,经0.22μm微孔滤膜过滤,按仪器工作条件进行测定。
2 结果与讨论
2.1 色谱行为
14种甾体激素在不同离子模式下的总离子流色谱图见图1。
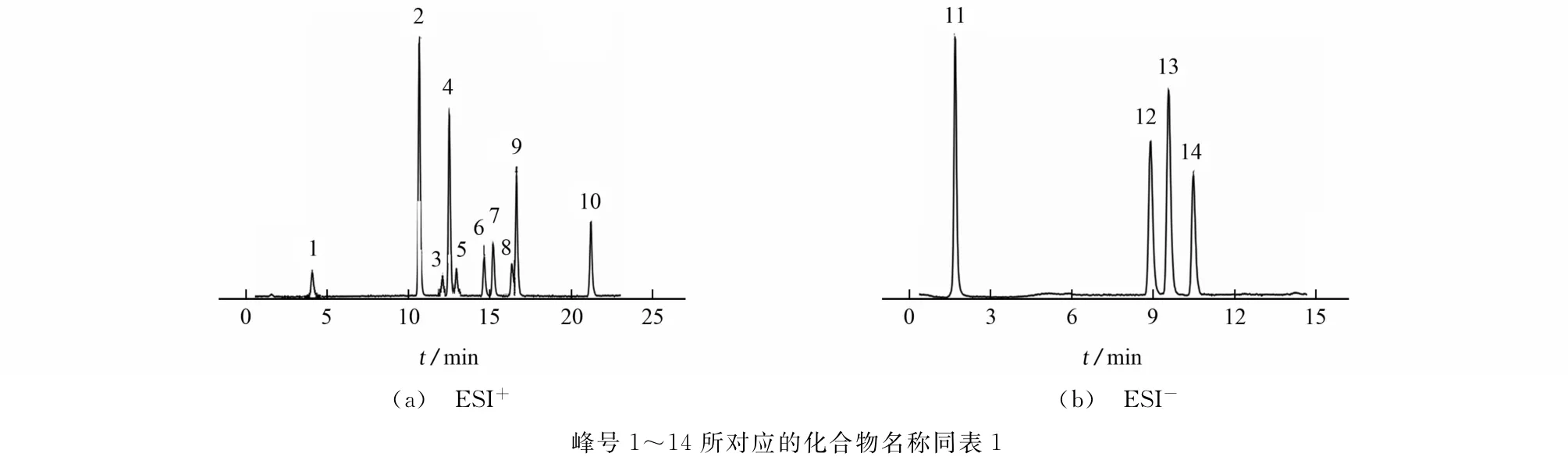
图1 14种甾体激素在不同离子模式下的总离子流色谱图Fig.1 TIC chromatograms of 14steroid hormones in different ion modes
2.2 提取剂的选择
试验考察了甲醇、乙腈、乙酸乙酯、乙醚、异丙醇、正己烷、石油醚等有机溶剂对尿液中甾体激素的提取效果。结果表明:不同有机溶剂对尿液中14种甾体激素的提取效率不同,其中乙醚对甾体激素的提取率较高,最高值95.4%,平均值84.9%;其次石油醚对甾体激素的提取率平均值为80.4%;而甲醇对甾体激素的提取率较低,最低值56.1%,平均值为75.9%,这可能是由于尿液基质中干扰物易溶于甲醇,从而降低了甲醇的提取效果。试验选择乙醚作为提取剂。
2.3 SPE净化柱的选择
通过空白基质加标试验,考察了不同SPE净化柱(C18、MCX、HLB、MAX、WAX、WCX、SCX)对甾体激素(以雌酮为例)的净化效果,见图2。
由图2可知:弱阳离子交换柱(HLB、WCX)和C18柱对甾体激素具有较好的萃取、分离、浓缩效果,尤其是HLB柱回收率高达96.7%;强阳离子交换柱(MCX和SCX)净化效果适中;而弱阴离子交换柱(MAX和WAX)净化效果较差,尤其是MAX柱回收率仅为41.2%。因此,试验选择HLB固相萃取柱作为净化柱。

图2 SPE净化柱对甾体激素回收率的影响Fig.2 Effect of SPE purification columns on recovery of steroids
2.4 色谱条件的选择
2.4.1 色谱柱
试验考察了 HSS T3、BEH C18、BEH HILIC等不同型号色谱柱对甾体激素分离效果的影响,比较其分离度和灵敏度。结果表明:HSS T3和BEH C18可以有效分离14种甾体激素,尤其HSS T3色谱柱(2.1mm×100mm,1.8μm)分离的色谱峰,峰形较窄、对称且尖锐,分离度大于1.5,同时HSS T3色谱柱耐受酸度范围较宽,能够满足酸性较强的流动相环境。试验选用HSS T3型色谱柱。
2.4.2 流动相
试验考察了不同比例的有机相(甲醇、乙腈)与水相(水、甲酸溶液、乙酸铵溶液)作为流动相时甾体激素分离的效果。结果表明:当有机相为甲醇,水相为水、甲酸溶液、乙酸铵溶液或含0.1%甲酸的乙酸铵溶液时,甾体激素分离度较差,峰宽且响应值较低;而当有机相为乙腈,水相为含0.1%甲酸的乙酸铵溶液时,甾体激素分离度较好,峰窄且尖锐,响应值较高,这可能是由于流动相中添加的甲酸,可以为甾体激素在电离过程中提供H+和COO-,使其获得较高丰度的[M+H]+或[M+HCOO]-分子离子峰。试验选择乙腈为有机相,含0.1%甲酸的乙酸铵溶液为水相。
2.5 质谱条件的选择
根据14种甾体激素的化学性质,分别采用1.0mg·L-1单标准溶液流动注射连续进样,流量为5.0μL·min-1,以正、负离子模式进行一级质谱扫描,确定准分子离子峰后进行二级质谱扫描,获得高丰度的特征碎片离子,结果见表1。除雌酮、17α-雌二醇、17β-雌二醇、雌三醇为负离子模式下获得[M-H]-准分子离子峰,其他10种甾体激素均为正离子模式下获得准分子离子峰,其中孕三醇获得[M+NH4]+的准分子离子峰,孕二醇因易脱水而获得[M-2H2O+H]+的准分子离子峰,其余为[M+H]+准分子离子峰。
2.6 基质效应
在乙腈、空白尿基质溶液中加入一定量的甾体激素混合标准溶液,配制成低(2.50μg·kg-1)、中(50.0μg·kg-1)、高(100.0μg·kg-1)等3个浓度水平溶液,按仪器工作条件进行测定。比较乙腈和尿基质溶液中被测组分的峰面积,计算基质效应(ME),ME=Ap/As×100%,其中Ap指尿基质溶液中被测组分的峰面积,As指纯乙腈溶液中被测组分的峰面积,结果见表2。
由表2可知:被测组分的基质效应在80.3%~99.2%之间,没有明显的基质效应。
2.7 标准曲线与检出限
在空白尿液基质中,分别加入适量的甾体激素混合标准溶液,配制成甾体激素质量分数依次为2.50,5.00,10.0,25.0,50.0,100.0μg·kg-1的混合标准溶液系列,按试验方法对其进行测定。以甾体激素的质量分数为横坐标,对应的色谱峰面积为纵坐标绘制标准曲线,其线性范围、线性回归方程和相关系数见表3。按3倍信噪比计算方法的检出限(3S/N),见表3。由表3可知:14种甾体激素的相关系数均大于0.999 0,线性关系较好;检出限在0.5~5.0μg·kg-1之间,说明方法灵敏度较高。

表3 线性参数和检出限Tab.3 Linearity parameters and detection limits
2.8 精密度和回收试验
在尿液空白基质中添加一系列质量分数的甾体激素混合标准溶液,按试验方法平行测定6次,精密度和回收试验结果见表4。
由表4可知:加标回收率在80.6%~99.2%之间,相对标准偏差(RSD)在1.1%~7.9%之间,说明方法的准确度和精密度较高。
2.9 样品分析
随机选取某医院泌尿外科待测实际尿液30份,按试验方法进行内源性甾体激素残留量的测定。结果表明:2份试样中检出孕酮,质量分数分别为6.3,2.8μg·kg-1;3份试样中检出表睾酮,质量分数在1.8~3.4μg·kg-1之间;5份试样中检出17β-雌二醇,质量分数在2.1~5.6μg·kg-1之间。说明尿液中存在一定量的内源性甾体激素残留。
本工作采用超高效液相色谱-串联四极杆飞行时间质谱法建立了人体尿液中14种内源性甾体激素(雌酮、17α-雌二醇、17β-雌二醇、雌三醇、丙酸睾酮、雄酮、本胆烷醇酮、表睾酮、脱氢表雄酮、睾酮、孕酮、孕二醇、孕三醇、氢化可的松)残留量的分析方法。方法准确度高、精密度高、检出限低,可为尿液中甾体激素的日常检测提供分析方法。
[1] 秦伟,刘桂华.动物源性食品中性激素残留检测方法研究进展[J].中国预防医学杂志,2010,11(7):746-749.
[2] 张国胜,董学芝,李畅,等.肉类食品激素残留检测技术研究进展[J].食品科学,2008,29(2):481-486.
[3] 王静竹.同位素比质谱法检测尿中类固醇来源[J].中国运动医学杂志,2006,25(6):725-728.
[4] ZHANG S J,YOU J M,NING S J,et al.Analysis of estrogenic compounds in environmental and biological samples by liquid chromatography-tandem mass spectrometry with stable isotope-coded ionization-enhancing reagent[J].J Chromatogr A,2013,1280:84-91.
[5] 王炼,杨元,王林.酶水解-高效液相色谱法测定肉类食品及牛奶中11种甾体激素[J].理化检验-化学分册,2007,43(6):482-484.
[6] SCHIPILLITI L,BONACCORSI I,COTRONEO A,et al.Evaluation of gas chromatography combustion-isotope ratio mass spectrometry (GC-C-IRMS)for the quality assessment of citrus liqueurs [J].Journal of Agricultural and Food Chemistry,2013,61(8):1661-1770.
[7] 罗庆,孙丽娜,杨悦锁,等.固相萃取-衍生化-气相色谱-质谱法测定水中类固醇类雌激素[J].理化检验-化学分册,2015,51(10):1406-1410.
[8] 张毅,岳振峰,蓝芳,等.分散固相萃取净化与液相色谱/串联质谱法测定牛奶中8类禁用药物残留[J].分析化学,2012,40(5):724-729.
[9] 贺利民,黄显会,方炳虎,等.超高效液相色谱-串联质谱法测定动物肌肉组织和鸡蛋中残留的11种甾体激素类药物[J].色谱,2008,26(6):714-719.
[10] 张鸿伟,蔡雪,林黎明,等.液相色谱-四极杆/离子阱质谱同时确证和测定肌肉中16种同化甾体激素残留[J].色谱,2012,30(10):991-1001.
