慢电子速度成像技术
2018-01-19罗志弘宁传刚
赵 静 罗志弘 宁传刚
(1北京航空航天大学仪器科学与光电工程学院,北京 100191; 2清华大学物理系,北京 100084)
0 引言
光电子能谱是物理化学领域研究物质结构和光电离动力学的重要工具。结合质谱技术的负离子光电子能谱仪,可以通过质谱选择特定的负离子进行光电子能谱的测量。负离子光电子谱仪的能量分辨率高,灵敏度高,可以对自由基、反应中间体、团簇等不易大量制备的体系开展研究。测量负离子光电子能谱,可以得到电子亲和势(Electron Affinity,EA)[1,2]和电子能级结构等物质元素的基本信息。对分子团簇,高分辨的光电子能谱还可以分辨出振动结构。这些信息可以帮助人们确定原子分子团簇的结构,理解其性质。特别是研究自由基和反应中间体这些短寿命体系,其他实验手段通常难以开展研究。负离子光电子能谱实验,最早由Hall和其合作者在1967年首次报道[3],目前已经发展成为原子分子物理和化学领域广泛使用的研究手段。
负离子光电子能谱仪的光源通常采用激光,对电子的动能测量早期采用静电型能量分析器,如半球形能量分析器,典型的能量分辨率约5meV,但光电子的收集效率较低[4-6]。后期,发展了收集效率接近100%的磁瓶式电子飞行时间测量装置[7,8],能量分辨率和半球形能量分析器相当。最近,新发展起来的慢电子速度成像技术可以实现好于0.2meV的能量分辨率[9]。利用线偏振激光做光源脱附负离子,产生的光电子具有旋转对称性。因此,对速度成像得到的二维电子分布进行反阿贝尔(反Abel)变换,就可以重建光电子分布,进而测量动能和角分布(photoelectron angular distribution,PAD)[10-12]。慢电子速度成像的基本思想非常简单,如果能保持相对能量分辨率ΔE/Ek=2%,对动能Ek=1000meV的电子,绝对能量分辨率ΔE=20meV;而对动能为Ek=10meV的电子,ΔE=0.2meV。低能电子的飞行轨迹特别容易受到杂散电磁场干扰,这使得常规的静电型分析器和磁瓶飞行时间谱仪的极限能量分辨率很难好于1meV。而慢电子速度成像测量方式对杂散的电磁场不敏感,因此对低能电子可以实现很高的能量分辨率。目前,我们课题组搭建的慢电子速度成像谱仪可以实现优于0.1meV的能量分辨率。利用慢电子速度成像技术,我们已经成功地将多个过渡族元素的电子亲和势测量精度提高了两个量级。
1 速度成像技术的发展
离子速度成像技术是最早由Chandler和Houston[13]等人于1987年引入,他们用这个方法测量了光解离产物的角分布,但能量分辨率很差。1997年,Eppink和Parker[14]等人对其进行了改进,通过增加一片聚焦离子透镜,使得能量分辨大大提高,典型的能量分辨率约为2%。在这之后,离子速度成像技术得到了广泛的应用。离子速度成像技术的基本原理是,分子束(或离子束)与一束线偏振激光垂直交叉,光解离(或光脱附)后的离子碎片(或光电子)在外加电场的作用下加速后,再经一段自由飞行,打在位置灵敏探测器上。常用的位置灵敏探测器由微通道板(MCP)和荧光屏组成。离子(或电子)打在荧光屏上的位置被CCD相机观测和记录下来。由于离子(或电子)速度分布具有旋转对称性,通过反阿贝尔变换可以重构出离子球(或电子球)的三维分布。
在最初的传统成像阶段,Chandler[13]等人的实验采用共振多光子选态电离,外加电场由简单的两片平板电极构成,如图1(a)所示。推进极LR和带栅网结构的引出极LE将离子加速,飞向荧光屏。这种两片式离子速度成像的分辨率主要受反应区尺寸、均匀电场导致的图像重叠,以及栅网结构对离子透过率影响等因素限制,能量分辨率非常有限。Eppink和Parker[14]等人改进了成像电极的结构。成像透镜由三片电极构成,引出极的栅网换成了中心有孔的极板,提高了离子透过率,从而增强信号强度。三片式结构的最大改进是可以将速度相同的离子碎片聚焦到探测器的同一点上,从而使实验分辨率得到了显著提升。速度成像的数据处理都需要进行反阿贝尔变换。在实验数据统计涨落较大时,反阿贝尔变换会产生很多噪声。为了克服这一缺点,Gebhardt[15]等人于2001年提出的切片成像技术中,先让离子球自由膨胀,通过延迟给膨胀的离子球上所加电场的时间,增加离子团信号到达MCP的峰宽,从而探测dx厚度的离子团切片信号,直接记录离子碎片的三维图像,避免了反阿贝尔变换带来的可能误差,也可以达到很高的分辨率。但由于电子质量轻,飞行速度快,该方法不适用,所以,切片技术目前只限于离子速度成像。
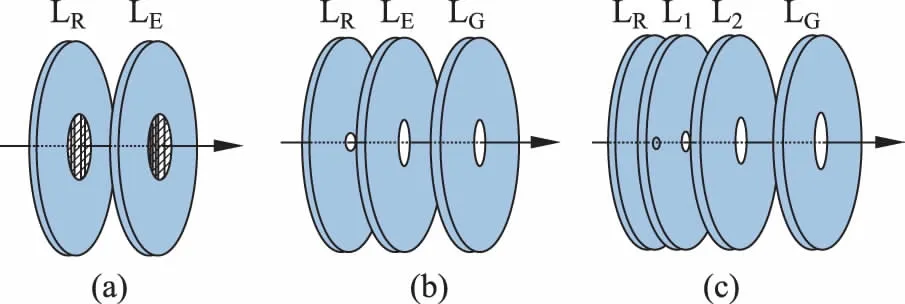
图1 离子透镜结构示意图(a) 两片栅网电极; (b) 三片电极; (c) 四片电极
将速度成像技术引入负离子光电子谱仪[16-24],为电子的能谱测量提供新的选择。速度成像的优点是光电子收集效率为100%,在测量电子能量的同时还可以得到光电子的角分布信息。
SEVI的光电子动能很低,一般约为10~20meV。零动能光电子谱(zero electron kinetic energy,ZEKE)[28]技术是在20世纪80年代中期由PES发展而来,其能量分辨率可达到0.1~0.2meV,适用于较小的负离子团簇。从某种意义上说,SEVI是传统PES与ZEKE相结合的产物。但是,由于Wigner阈值定理[29],光脱附截面σ∝(Ek)l+1/2。这里l为出射光电子的角动量。因此,光电子动能越低,高分波光脱附截面下降得也越快,在阈值附近,高分波通道信噪比变得很差。这使得ZEKE仅限于测量l=0的s分波光脱附通道。
最近,王来生小组对三片式成像电极做了进一步改进[30]。参考离子成像的四片式结构[31],新设计的四片式透镜的聚焦性能更好。对阈值慢电子能量分辨率达到了1.2cm-1,对较大动能的电子可以实现ΔE/Ek=0.53%。我们实验室采用的正是这种四片式电极。
2 慢电子速度成像谱仪系统结构
离子速度成像实验装置一般由离子源、飞行时间质谱系统、离子探测系统、信号采集系统组成,各部分均置于真空腔体内。为了减小样品损耗,提高使用效率,同时减小分子泵负载,提高装置内的动态真空,离子的产生和探测采用脉冲方式,由脉冲时序控制器进行统一的时序控制。我们的离子源采用激光溅射离子源。将脉冲Nd:YAG激光器的二次谐波(532nm)聚焦成直径约1mm的光斑并打到样品靶上,在样品靶表面产生包含各种中性和带电离子的激光诱导等离子体。同时脉冲阀向等离子体区域喷入氦气作为载气,载气一边与等离子体混合对其进行冷却,一边与各种粒子一起以超音速向前膨胀扩散。粒子团簇和氦气的混合物经一个漏斗状滤束器(skimmer)二次筛选,滤去横向速度大的粒子,使最初向各个方向扩散的混合物变成一束较为平直的束流,进入飞行质谱仪的加速区。飞行时间质谱仪筛选出感兴趣的负离子。离子束被一系列偏转板导引、聚焦,经过一段时间的无场自由飞行后到达飞行时间质谱仪末端的离子质量门。质量门由三片式栅网电极组成,前后两片电极接地,中间电极加脉冲高压,实现对离子的“开”和“关”。不同荷质比的粒子飞行时间不同,通过改变质量门的开门时间,允许指定离子通过质量门。离子探测器是由两片级联的微通道板(MCP)和一片金属板构成。离子探测可以转动,在进行质谱测量时,离子探测器与离子束共线。在进行光电子谱测量时,需要将离子探测器从离子飞行路径中转开。
在成像透镜工作区,离子束与脱附激光垂直交叉。脱附激光是窄线宽的可调谐染料激光器。染料激光器为Nd:YAG脉冲激光器泵浦。染料激光器的调谐范围为400~920nm,线宽为0.06cm-1。光子能量(hν)用高精度的波长计(WS6-600)进行测量。波长计的精度为0.02cm-1。脱附后的光电子在电场的加速下向荧光屏飞行。目前国内没有大尺寸快速荧光屏商品,我们自制了直径75mm的荧光屏(灵敏面积)。它由两块直径80mm的微通道板和快荧光板构成。光电子经微通道板倍增106~107倍,产生的大量二次电子被电场加速(3kV)并轰击荧光板,产生的荧光被CCD相机记录。由于荧光肉眼可见,对CCD相机要求不高。每个电子轰击荧光屏产生的亮斑大小在1mm左右,直接累计这些信号,亮斑的大小就成了限制能量分辨的主要因素。可以通过对各个像素加权求重心的方式精确定位光电子的位置。利用求重心的方法还可以比较容易对图像的畸变进行实时修正。速度成像的分辨率和离子束-激光束的交叉重叠区的大小密切相关。交叉区越小,分辨率越好。但交叉区太小,信号强度会损失很多。在我们的装置中,激光束通过3mm的准直模孔与离子束交叉。成像透镜的第一级有一直径为6mm的孔,用来限制交叉区离子束尺寸。此外,要得到好的能量分辨率,各个电极的分压需要仔细调节,离子束和激光束的交叉重叠位置也需要很好优化。由于电子散落在1024×1024像素大小的二维平面上,每张能谱一般需要累积50000~200000发激光脉冲[32],以尽可能降低计数统计涨落。
3 能量定标与数据处理
以原子负离子被光脱附过程为例。负离子被激光脱附掉一个电子,对于来自同一电子态的光电子,它们有相同的动能,这些光电子经无场自由飞行后会膨胀成一个半径为r的球壳。到达探测器时,由三维球壳分布投影成一张二维的速度分布图像。速度分布图像呈一个半径为r的圆环分布,如图2所示。
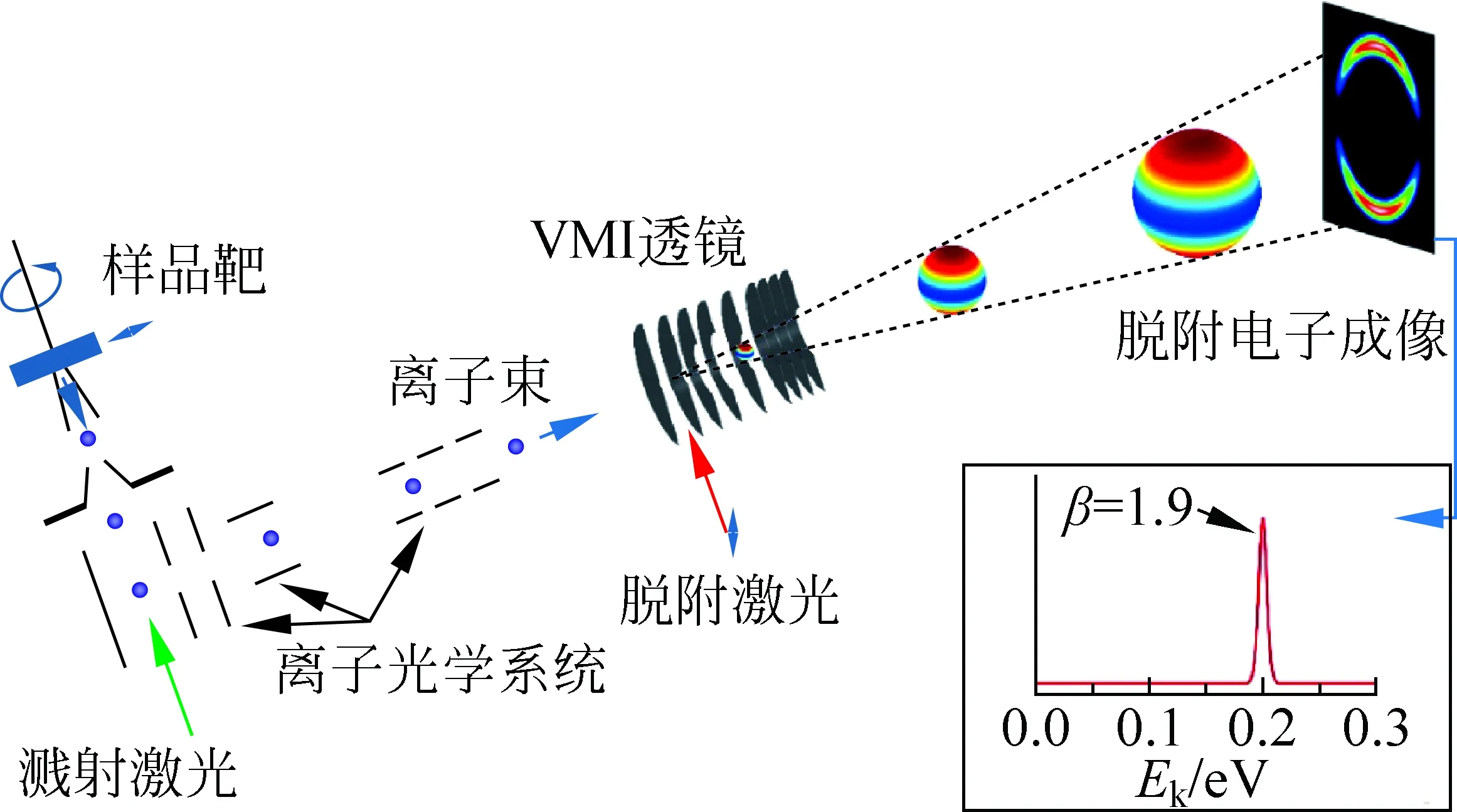
图2 SEVI原理图
在电子被光脱附后的飞行过程中,电子球壳半径r=vt。这里t是电子飞行时间,v为飞行速度。因此,电子动能Ek可由下式给出:
αr2
(1)
相应地,该电子态的结合能BE=hν-αr2。电子动能Ek和球壳的半径r2成正比。通过改变脱附光子能量hν,测量一系列点,就可以得到能量定标系数α。
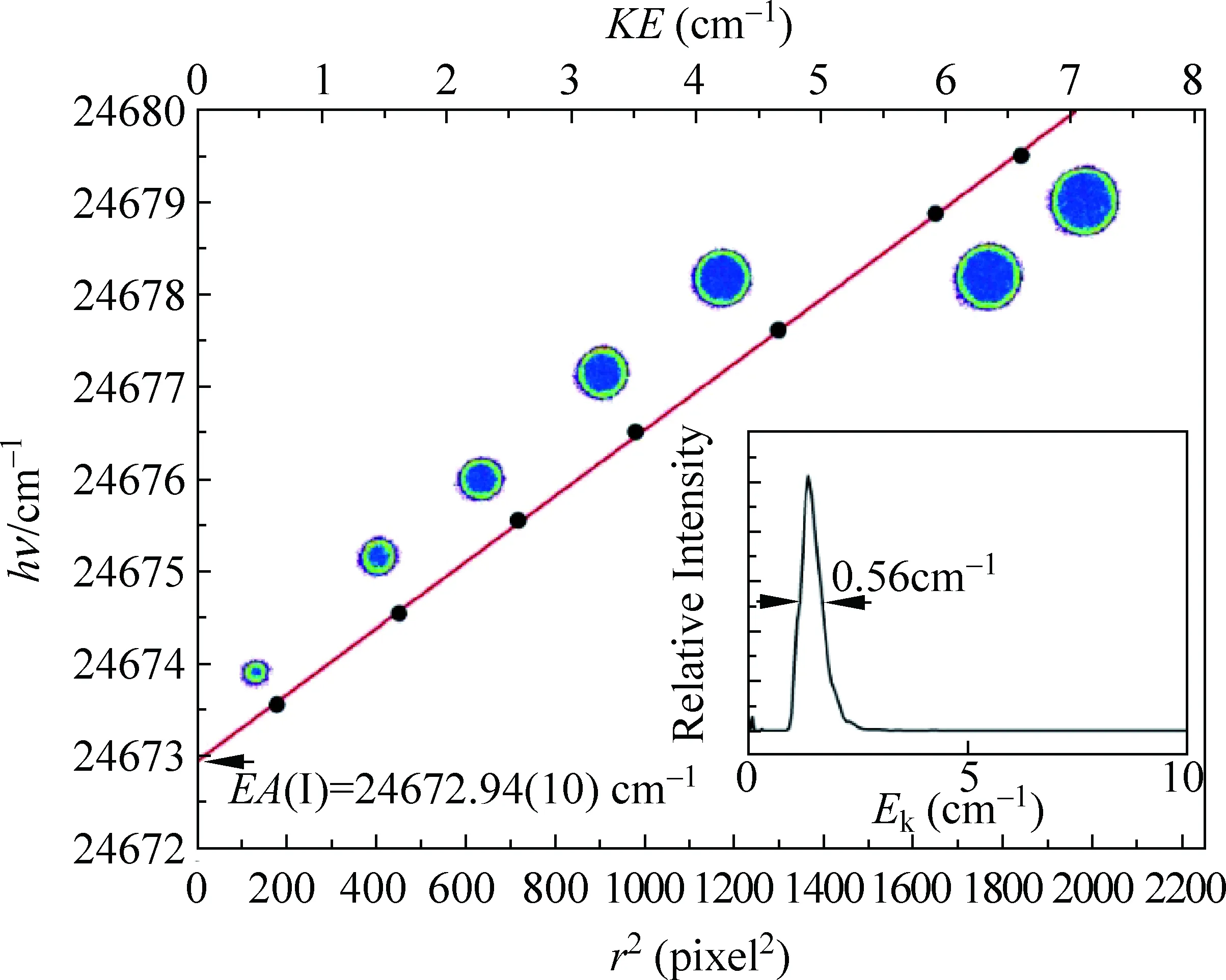
图3 用I-进行能量定标图
碘负离子电子亲和势较高,很容易得到很强的质谱信号,因此常用来作为系统的定标元素。图3为我们组搭建的SEVI仪器用碘负离子定标结果。成像电压是-150V。在一系列略高于I-脱附阈值的激光能量下分别进行实验,采集数据并处理后获得图中7个数据点。横坐标为CCD相机采集到圆环半径的平方r2,纵坐标为光子能量hν。可以看到,图像中圆环的半径随激光能量的增加而不断增大。对7个数据点进行线性拟合,可获得仪器的能量定标系数α。线性拟合得到的截距(r=0处)就是碘的电子亲和势。用这个方法,我们得到的碘电子亲和势为24672.94cm-1,和用LPM结果3.059038(10)eV[33]符合得很好。图3中的小插图显示的是动能为1.4cm-1时的能谱,峰的半高全宽(FWHM)为0.56cm-1。这是目前报道的最窄峰宽。
脱附光源为线偏振光,且线偏振方向和荧光屏平行。因此,出射的光电子的分布具有旋转柱对称性。光电子的动能和角分布可以全部从投影的二维速度成像中重建出来,不会损失任何信息。
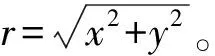
(2)
根据阿贝尔变换公式,有
(3)
通过反阿贝尔变换可以由原始二维图像fx得出电子速度分布fr,z0
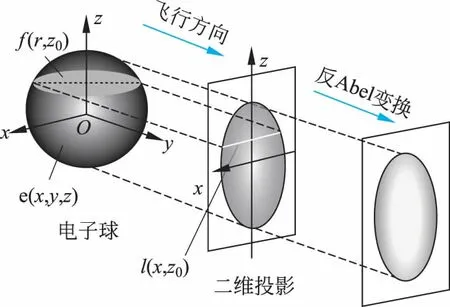
图4 反阿贝尔变换原理图
(4)
目前,利用二维速度成像结果重建电子能谱和角分布已经有不少共享程序可以实现。常用软件有BASEX[34],pBasex[35],Onion Peeling[36],Polar Onion Peeling[37],以及Meveler[38]。Iker León等人对比过以上几种方式,大多数情况下它们处理结果没有明显差异[30]。但BASEX可以自由选择步长,因此对数据的处理更为灵活;pBasex利用了单光子脱附的全部特征信息,而Basex只用了柱对称性,因此pBasex可以给出更为平滑的曲线,更好地抹平统计涨落,但分辨率有所损失;Onion Peeling方法在计算指定峰的各向异性参数上更有优势;Polar Onion Peeling方法是用LABVIEW编写,所以和同为LABVIEW编写的图像采集软件有更好的适配性。Meveler在计数较少时,依然能给出较好的变换结果。
对于线偏振单光子光脱附,光电子的角分布由下式给出[12]
(5)
式中,θ是光电子出射方向相对于激光偏振方向的夹角。σtotal是光脱附总截面,β是各向异性参数,P2(cosθ)是二阶Legendre多项式。β取值范围在-1和2之间,具体数值和电子态的对称性以及电子动能有关。如果β是正值,意味光电子在平行偏振方向的强度比垂直方向更强。例如,光脱附一个s子壳层的电子产生p波,β为正值。这也是图2所示意的情形。如果β为负值,在垂直偏振方向上的光电子强度比平行方向更强。这通常是由于光脱附产生的光电子不同分波之间干涉的结果。例如,光脱附O-负离子的一个p电子,可以得到s分波和d分波,在一定动能下,β为-1。
4 应用
近年来,慢电子速度成像法以其超高能量分辨率在原子分子领域取得了许多令人瞩目的成果。例如,最近我们利用慢电子速度成像方法大大提高了多个过渡族元素电子亲和势的测量精度。电子亲和势是衡量原子得到电子容易程度的一个基本参数。目前,元素周期表中主族元素的电子亲和势的测量精度都比较高。这些数据基本上都是通过激光阈值光脱附谱学(LPT)和激光光脱附显微镜(LPM)这两种方法得到的[33,39]。由于过渡族元素独特的电子结构,在阈值附近的光脱附电子为p波。根据Wigner阈值定理,光脱附截面非常小,又由于精细结构造成多条能量间隔很小的跃迁通道都有机会打开,这使得传统的测量电子亲和势LPT和LPM方法无能为力。目前,已报道的过渡族元素电子亲和势大多数是由Lineberger等人早期用静电型能量分析器PES方法得到。其测量精度不高,不确定度约10meV,且近30年无明显提高[4]。测量过渡族元素的电子亲和势的另一个难点在于,过渡族原子的电子亲和势很低,又特别容易氧化,使用通常的激光溅射离子源,信号主要是氧化物的负离子信号,单原子负离子的信号非常弱。
2015年,我们报道了自行搭建的一套高精度SEVI装置,并首次将SEVI方法应用在过渡族原子负离子电子结构研究中,精确测定了元素周期表中多个过渡族元素的电子亲和势,如铌(Nb)[40],钴(Co)[32],铅(Pb)[41],锆(Zr)[42],铼(Re)[43]等,使过渡族元素EA值的测量精度有了极大的提高。其中,Nb的EA值测量精度提高了400多倍。对于Re,长期以来人们一直认为其和同族的Mn-负离子一样,不能稳定存在。我们的实验明确无误地表明Re-负离子稳定存在,并精确测量了电子亲和势。利用SEVI我们不仅可以得到电子亲和势,还得到了负离子的精细结构。过渡族元素是很多功能材料的关键成分,如磁性、超导、催化剂等。但是由于电子关联的多体效应,过渡族元素的理论计算一直是一个挑战。我们测量得到的高分辨光电子谱将为发展可以处理过渡族化合物的新理论方法提供检验标准。
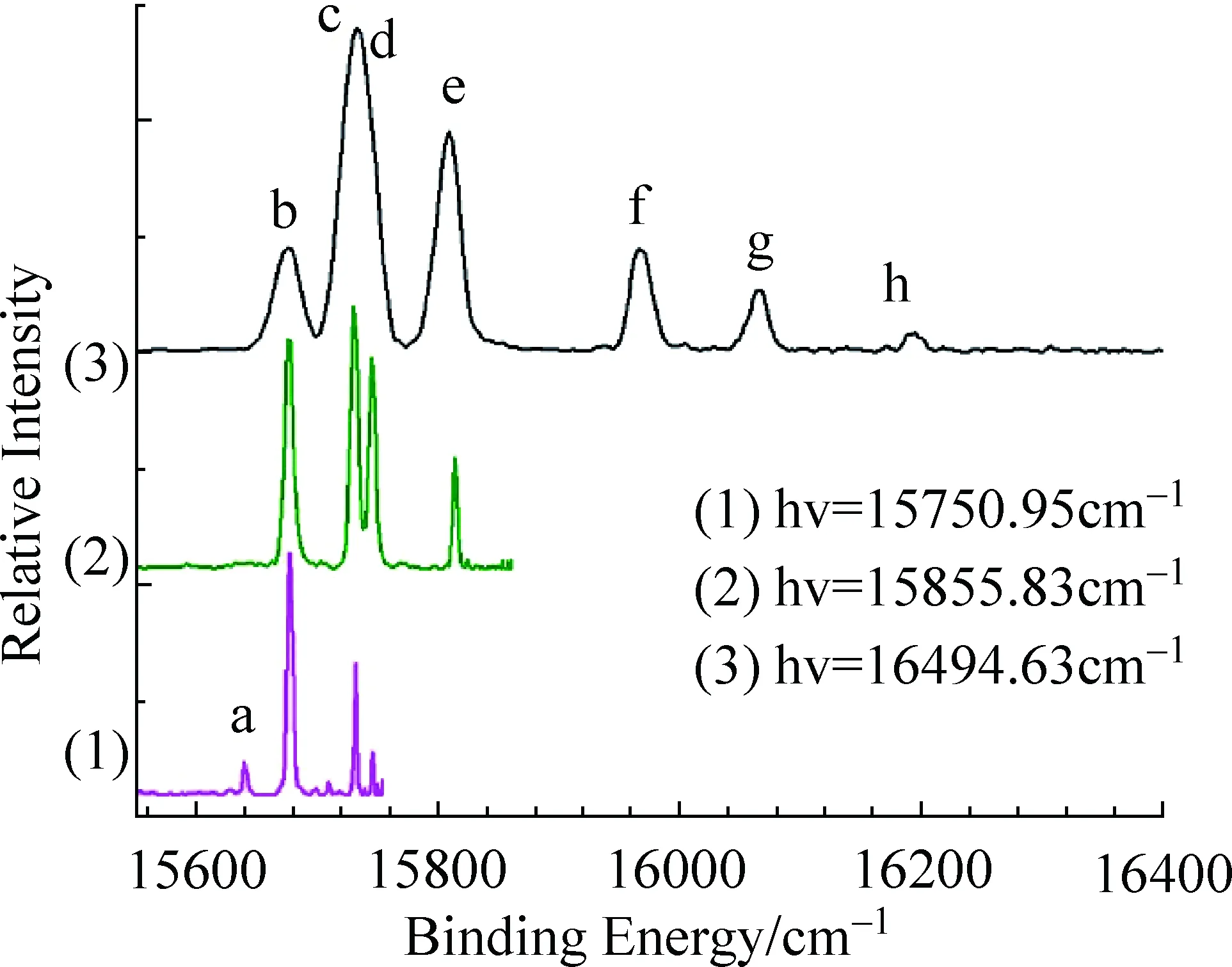
图5 不同激光能量下脱附所采集的Nb-光电子谱图
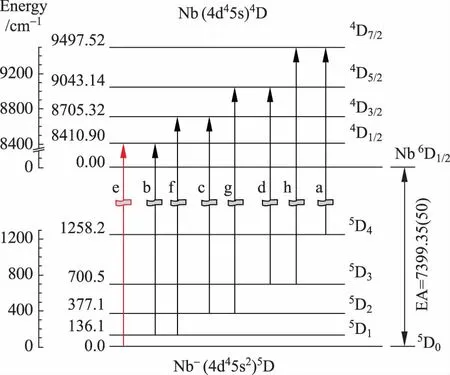
图6 Nb和Nb-间跃迁能级图
图5是用SEVI方法得到的[40]铌(Nb-)光电子能谱。脱附激光能量分别为15750.95cm-1,15855.83cm-1与16494.63cm-1。图5中不同字母标识出的峰分别对应图6中的不同跃迁;图5中不同颜色的光电子谱分别对应不同能量的脱附激光。可以看出,随着光电子动能的降低,相邻的峰c和d被逐渐分开。这很好展示了能量分辨率随光电子动能的降低而逐渐提高。峰e对应Nb-负离子基态5D0到Nb中性原子4D1/2的跃迁。进行如图3所示的测量,在通道e脱附阈值之上20cm-1到60cm-1区间测量一系列能谱,就可以得到峰e的精确结合能。由于中性原子4D1/2能级有很精确的光谱数据,从测量得到的结合能15810.25(50)cm-1中减去4D1/2的能级8410.90cm-1,就得到Nb的电子亲和势7399.35(50)cm-1或917.40(6)meV。这一结果同早期报道的894(25)meV一致,但精度提高了400多倍。由于可以分辨多个跃迁通道,测量电子亲和势不一定限于跃迁到中性原子基态的通道,这样可以避免使用不易获得的可调谐窄线宽红外宽激光器。例如,如果直接测量Nb-(5D0)→Nb(4D1/2)跃迁通道的结合能(结合能7399.35cm-1),就需要输出波长在1351nm左右的可调谐红外激光器。这个特点对测量电子亲和势很低的元素尤其重要,例如Re的电子亲和势为60.396(63)meV[43]。
SEVI的超高能量分辨率使得其可以分辨分子体系的振动结构。但对分子体系的测量,由于室温下分子振动、转动的存在,造成谱线的热展宽。由于大分子的转动间隔一般小于0.1cm-1,分辨率不足以分辨转动结构。2013年,Lai-Sheng Wang组将SEVI与冷离子阱技术(cold ion trap)相结合,大大降低了谱线的热展宽[44-50]。例如,他们将脱氢苯酚负离子(C6H5O-)降温至20K(低温下分子负离子处于振动基态,转动主要布居在较低能态)实现了1meV的能量分辨率,并分辨了很多振动频率低至~100cm-1的低频振动模式[50]。Neumark组也报道过一系列分子体系的光电子能谱实验研究,如燃烧过程中蒽基自由基[51],亚乙烯基(H2CC-)[52,53]等。最近,他们利用这个方法还观测到了F+H2反应中的共振过渡态[54]。
5 总结与展望
慢电子速度成像方法是新发展起来一项光电子谱技术,它可以实现优于1cm-1的能量分辨率,结合质谱和冷离子阱技术,可以预期其在原子分子化学物理领域得到更广泛的应用。其特点总结如下(1)质谱选择特定质量的离子;(2)具有接近100%的收集和探测效率,灵敏度高,效率高,且能获得光电子角分布等信息;(3)对杂散电磁场不敏感,对多普勒效应不敏感,具有很高的能量分辨率;(4)实验所需样品量小,有利于对放射性元素或稀有元素的研究;(5)与离子阱冷却相结合,能有效降低大分子的振动转动热展宽,减小激发态离子布居,降低实验复杂度。慢电子速度成像方法所提供的高分辨谱学数据将为发展高精度的化学反应动力学以及过渡族多体问题的新理论新方法提供可靠的检验数据。可以预期,凭借其超高的能量分辨率,结合冷离子阱技术的慢电子速度成像方法将会得到越来越广泛的应用。
[1] Rienstra-Kiracofe J C, Tschumper G S, Schaefer H F, et al. Atomic and molecular electron affinities: photoelectron experiments and theoretical computations[J]. Chemical Reviews, 2002, 102(1): 231-282.
[2] Branscomb L M, Burch D S, Smith S J, et al. Photodetachment cross section and the electron affinity of atomic oxygen[J]. Physical Review, 1958, 111(2): 504.
[3] Brehm B, Gusinow M A, Hall J L. Electron affinity of helium via laser photodetachment of its negative ion[J]. Physical Review Letters, 1967, 19(13): 737.
[4] Feigerle C S, Corderman R R, Bobashev S V, et al. Binding energies and structure of transition metal negative ions[J]. The Journal of Chemical Physics, 1981, 74(3): 1580-1598.
[5] Ervin K M, Lineberger W C. Photoelectron spectra of dicarbon and ethynyl [J]. The Journal of Physical Chemistry, 1991, 95(3): 1167-1177.
[6] Breyer F, Frey P, Hotop H. High resolution photoelectron spectrometry of negative ions: Rotational transitions in laser-photodetachment of OH-, SH-, SD-[J]. Zeitschrift für Physik A Atoms and Nuclei, 1981, 300(1): 7-24.
[7] Kruit P, Read F H. Magnetic field paralleliser for 2π electron-spectrometer and electron-image magnifier[J]. Journal of Physics E: Scientific Instruments, 1983, 16(4): 313.
[8] Wang L S, Cheng H S, Fan J. Photoelectron spectroscopy of size-selected transition metal clusters: Fe-n, n=3-24[J]. The Journal of Chemical Physics, 1995, 102(24): 9480-9493.
[9] Neumark D M. Slow electron velocity-map imaging of negative ions: Applications to spectroscopy and dynamics[J]. The Journal of Physical Chemistry A, 2008, 112(51): 13287-13301.
[10] Cooper J, Zare R N. Angular distribution of photoelectrons[J]. The Journal of Chemical Physics, 1968, 48(2): 942-943.
[11] Ning C G, Dau P D, Wang L S. Guiding Electron Emissions by Excess Negative Charges in Multiply Charged Molecular Anions[J]. Physical Review Letters, 2010, 105(26): 263001.
[12] Liu Y, Ning C G. Calculation of photodetachment cross sections and photoelectron angular distributions of negative ions using density functional theory[J]. The Journal of Chemical Physics, 2015, 143(14): 144310.
[13] Chandler D W, Houston P L. Two-dimensional imaging of state-selected photodissociation products detected by multiphoton ionization[J]. The Journal of Chemical Physics, 1987, 87(2): 1445-1447.
[14] Eppink A T J B, Parker D H. Velocity map imaging of ions and electrons using electrostatic lenses: Application in photoelectron and photofragment ion imaging of molecular oxygen[J]. Review of Scientific Instruments, 1997, 68(9): 3477-3484.
[15] Gebhardt C R, Rakitzis T P, Samartzis P C, et al. Slice imaging: A new approach to ion imaging and velocity mapping[J]. Review of Scientific Instruments, 2001, 72(10): 3848-3853.
[16] Deyerl H J, Alconcel L S, Continetti R E. Photodetachment imaging studies of the electron affinity of CF3[J]. The Journal of Physical Chemistry A, 2001, 105(3): 552-557.
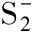
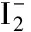
[19] Rathbone G J, Sanford T, Andrews D, et al. Photoelectron imaging spectroscopy of Cu-(H2O) 1, 2 anion complexes[J]. Chemical Physics Letters, 2005, 401(4): 570-574.
[20] Sobhy M A, Castleman Jr A W. Photoelectron imaging of copper and silver mono-and diamine anions[J]. The Journal of Chemical Physics, 2007, 126(15): 154314.
[21] McCunn L R, Gardenier G H, Guasco T L, et al. Probing isomer interconversion in anionic water clusters using an Ar-mediated pump-probe approach: Combining vibrational predissociation and velocity-map photoelectron imaging spectroscopies[J]. The Journal of Chemical Physics, 2008, 128(23): 234311.
[22] Bartels C, Hock C, Huwer J, et al. Probing the angular momentum character of the valenceorbitals of free sodium nanoclusters[J]. Science, 2009, 323(5919): 1323-1327.
[23] Wu X, Qin Z, Xie H, et al. Vibrationally resolved photoelectron imaging of gold hydride cluster anions: AuH-and Au2H-[J]. The Journal of Chemical Physics, 2010, 133(4): 044303.
[24] Liu Q Y, Hu L, Li Z Y, et al. Photoelectron imaging spectroscopy of MoC-and NbN-diatomic anions: A comparative study[J]. The Journal of Chemical Physics, 2015, 142(16): 164301.
[25] Osterwalder A, Nee M J, Zhou J, et al. High resolution photodetachment spectroscopy of negative ions via slow photoelectron imaging[J]. The Journal of Chemical Physics, 2004, 121(13): 6317-6322.
[26] Cavanagh S J, Gibson S T, Gale M N, et al. High-resolution velocity-map-imaging photoelectron spectroscopy of the O-photodetachment fine-structure transitions[J]. Physical Review A, 2007, 76(5): 052708.
[27] Hock C, Kim J B, Weichman M L, et al. Slow photoelectron velocity-map imaging spectroscopy of cold negative ions[J]. The Journal of Chemical Physics, 2012, 137(24): 244201.
[28] Fischer I, Lindner R, Müller-Dethlefs K. State-to-state photoionisation dynamics probed by zero kinetic energy (ZEKE) photoelectron spectroscopy[J]. Journal of the Chemical Society, Faraday Transactions, 1994, 90(17): 2425-2442.
[29] Wigner E P. On the behavior of cross sections near thresholds[J]. Physical Review, 1948, 73(9): 1002.
[30] León I, Yang Z, Liu H T, et al. The design and construction of a high-resolution velocity-map imaging apparatus for photoelectron spectroscopy studies of size-selected clusters[J]. Review of Scientific Instruments, 2014, 85(8): 083106.
[31] Townsend D, Minitti M P, Suits A G. Direct current slice imaging[J]. Review of Scientific Instruments, 2003, 74(4): 2530-2539.
[32] Chen X, Ning C. Accurate electron affinity of Co and fine-structure splittings of Co-via slow-electron velocity-map imaging[J]. Physical Review A, 2016, 93(5): 052508.
[33] Blondel C, Delsart C, Dulieu F. The photodetachment microscope[J]. Physical Review Letters, 1996, 77(18): 3755.
[34] Dribinski V, Ossadtchi A, Mandelshtam V A, et al. Reconstruction of Abel-transformable images: The Gaussian basis-set expansion Abel transform method[J]. Review of Scientific Instruments, 2002, 73(7): 2634-2642.
[35] Garcia G A, Nahon L, Powis I. Two-dimensional charged particle image inversion using a polar basis function expansion[J]. Review of Scientific Instruments, 2004, 75(11): 4989-4996.
[36] Manzhos S, Loock H P. Photofragment image analysis using the Onion-Peeling Algorithm[J]. Computer Physics Communications, 2003, 154(1): 76-87.
[37] Roberts G M, Nixon J L, Lecointre J, et al. Toward real-time charged-particle image reconstruction using polar onion-peeling[J]. Review of Scientific Instruments, 2009, 80(5): 053104.
[38] Dick B. Inverting ion images without Abel inversion: maximum entropy reconstruction of velocity maps[J]. Physical Chemistry Chemical Physics, 2014, 16(2): 570-580.
[39] Vandevraye M, Drag C, Blondel C. Electron affinity of selenium measured by photodetachment microscopy[J]. Physical Review A, 2012, 85(1): 015401.
[40] Luo Z, Chen X, Li J, et al. Precision measurement of the electron affinity of niobium[J]. Physical Review A, 2016, 93(2): 020501.
[41] Chen X, Ning C. Accurate electron affinity of Pb and isotope shifts of binding energies of Pb-[J]. The Journal of Chemical Physics, 2016, 145(8): 084303.
[42] Fu X, Li J, Luo Z, et al. Precision measurement of electron affinity of Zr and fine structures of its negative ions[J]. The Journal of Chemical Physics, 2017, 147(6): 064306.
[43] Chen X, Ning C G. Observation of Rhenium anion and electron affinity of Re[J]. The Journal of Physical Chemistry Letters, 2017, 8(12).
[44] Huang D L, Liu H T, Ning C G, et al. Resonant photoelectron imaging of deprotonated uracil anion via vibrational levels of a dipole-bound excited state[J]. Chemical Physics, 2017, 482: 374-383.
[45] Huang D L, Liu H T, Ning C G, et al. Conformation-Selective Resonant Photoelectron Spectroscopy via Dipole-Bound States of Cold Anions[J]. The Journal of Physical Chemistry Letters, 2015, 6(12): 2153-2157.
[46] Huang D L, Liu H T, Ning C G, et al. Vibrational state-selective autodetachment photoelectron spectroscopy from dipole-bound states of cold 2-hydroxyphenoxide: o-HO (C6H4) O-[J]. The Journal of Chemical Physics, 2015, 142(12): 124309.
[47] Liu H T, Huang D L, Liu Y, et al. Vibrational state-selective resonant two-photon photoelectron spectroscopy of AuS-via a spin-forbidden excited state[J]. The Journal of Physical Chemistry Letters, 2015, 6(4): 637-642.
[48] Huang D L, Liu H T, Ning C G, et al. Probing the vibrational spectroscopy of the deprotonated thymine radical by photodetachment and state-selective autodetachment photoelectron spectroscopy via dipole-bound states[J]. Chemical Science, 2015, 6(5): 3129-3138.
[49] Liu H T, Ning C G, Huang D L, et al. Vibrational Spectroscopy of the Dehydrogenated Uracil Radical by Autodetachment of Dipole-Bound Excited States of Cold Anions[J]. Angewandte Chemie International Edition, 2014, 53(9): 2464-2468.
[50] Liu H T, Ning C G, Huang D L, et al. Observation of Mode-Specific Vibrational Autodetachment from Dipole-Bound States of Cold Anions[J]. Angewandte Chemie International Edition, 2013, 52(34): 8976-8979.
[51] Weichman M L, DeVine J A, Levine D S, et al. Isomer-specific vibronic structure of the 9-, 1-, and 2-anthracenyl radicals via slow photoelectron velocity-map imaging[J]. Proceedings of the National Academy of Sciences, 2016, 113(7): 1698-1705.
[52] DeVine J A, Weichman M L, Zhou X, et al. Non-Adiabatic Effects on Excited States of Vinylidene Observed with Slow Photoelectron Velocity-Map Imaging[J]. Journal of the American Chemical Society, 2016, 138(50): 16417-16425.
[53] J A DeVine, M L Weichman, B Laws, et al. Encoding of vinylidene isomerization in its anion photoelectron spectrum[J]. Science, 2017, 358(6361): 336-339.
[54] Kim J B, Weichman M L, Sjolander T F, et al. Spectroscopic observation of resonances in the F+H2reaction[J]. Science, 2015, 349(6247): 510-513.
