液液萃取-气质联用法测定食用植物油中的敌草快含量
2018-01-19古丽君蓝康华陈玉浩阳洪波深圳市计量质量检测研究院广东深圳518100
古丽君,蓝康华,黄 磊,陈玉浩,阳洪波,唐 俗(深圳市计量质量检测研究院,广东 深圳 518100)
敌草快(Diquat)又名杀草快、双快,中文商品名为利农,化学名为1,1′-乙撑-2,2′-联吡啶二溴盐,分子式为C12H12N2Br2。敌草快是一种非选择性除草剂,可以终止植物光合作用,使植物迅速失水、枯萎死亡。敌草快对哺乳动物毒性大,摄入后对生物体的氧化还原活性影响很大,对肺、心、肝、肾等都有害,因此敌草快作为广谱性除草剂对环境的影响以及对人体健康的毒害越来越为人们所关注[1]。目前,文献报道过粮谷(玉米、大麦)、茶叶、水、血浆、尿液中敌草快含量检测方法,有高效液相色谱法[1-3]、高效液相色谱串联质谱法[4-7]、分光光度法[8-9]以及气相色谱串联质谱法[10-15]。本实验根据敌草快的理化性质,用水提取后,利用硼氢化钠将其还原为部分氢化产物,降低其沸点,再用气相色谱串联质谱进行定性定量分析。
1 材料与方法
1.1 实验材料
菜籽油、大豆油、玉米油,均为深圳市售。硼氢化钠(含量>96%);氢氧化钠(分析纯);二氯甲烷(分析纯);敌草快标准品(纯度99.5%), Dr. Ehrenstorfer GmbH;去离子水。
GC-MS-QP2010 Plus气相色谱-质谱联用仪(日本岛津),Sigma 3K15高速离心机,Genius 3 IKA Vortex涡旋混匀器。
1.2 实验方法
1.2.1 样品处理
提取:称取试样1 g(精确至0.01 g)置于15 mL塑料离心管中,加入1 mL二氯甲烷、1 mL去离子水,涡旋3 min后,于7 800 r/min离心3 min,用滴管将上层水相转移至另一塑料离心管中,有机层再加1 mL去离子水萃取,合并水层。
还原:上述提取液中加入20 mg 硼氢化钠,旋紧盖子,快速涡旋30 s混匀,室温下放置30 min。
还原产物提取:加入5 mol/L氢氧化钠溶液1 mL,再准确加入1 mL正己烷,涡旋3 min,4 500 r/min离心3 min,取出正己烷层,用适量无水硫酸钠干燥,过0.22 μm有机滤膜后,供气相色谱-质谱分析。
1.2.2 气相色谱条件
色谱柱:SH-Rxi-5sil MS毛细管柱(30 m×250 mm×0.25 μm);柱温60℃保持2 min,然后以10℃/min程序升温至180℃,再以30℃/min升温至300℃,保持3 min;载气为氦气(纯度≥99.999%),流速2.14 mL/min;进样口温度250℃;进样量1 μL;进样方式为无分流进样。
1.2.3 质谱条件
电子轰击源70 eV;离子源温度230℃;接口温度280℃;溶剂延迟时间2 min。选择性离子监测(SIM):m/z108、135、189、190。
1.2.4 敌草快含量的计算
试样中敌草快含量按下式计算:
X=(c-c0)×V×f/m
式中:X为试样中敌草快含量,mg/kg;c为样品测定液中敌草快的质量浓度,mg/L;c0为空白阴性试样敌草快的质量浓度,mg/L;V为样液最终定容体积,mL;f为稀释倍数;m为样品称样量,g。
2 结果与讨论
2.1 敌草快的质谱图和色谱图
敌草快经过硼氢化钠还原反应,转化成能被气相色谱所测定的物质。图1、图2分别是在空白大豆油中添加最低水平(0.18 mg/kg)的敌草快标准溶液,按1.2.1步骤处理后测得的全扫描质谱图和SIM模式扫描色谱图。

图1 敌草快全扫描质谱图

图2 敌草快SIM模式扫描色谱图
由图2可知,采用m/z108作为定量离子。
2.2 方法优化
2.2.1 提取过程优化
敌草快为季胺盐化合物,难溶于非极性有机溶剂,微溶于乙醇和羰基溶剂,易溶于水。本方法采用水作为提取溶剂,但水和植物油样品混合时易发生乳化,所以在提取时加入二氯甲烷,一方面可以避免乳化现象;另一方面可以使水层在液液萃取时处在上层,方便提取;同时,二氯甲烷还具有一定的净化作用。
2.2.2 反应时间优化
考察了5、10、15、20、30、40、50 min不同时间下还原效果,结果见图3。

图3 反应时间优化结果
从图3可以看出,反应时间为30 min时,敌草快还原产物的峰面积达到最大。因此,选择反应时间为30 min。
2.2.3 还原产物提取优化
硼氢化钠在碱性条件下稳定,在用正己烷提取敌草快还原产物前,加入适量的氢氧化钠溶液,可以中止剩余的硼氢化钠与水继续反应生成氢气,有利于还原产物的提取。
2.3 标准曲线
敌草快标准储备液的配制:精密称取敌草快标准品10 mg于100 mL容量瓶中,用少量去离子水溶解并稀释至刻度,即得质量浓度100 μg/mL标准储备液,4℃保存。
用去离子水将100 μg/mL标准储备液逐级稀释配制成质量浓度分别为0.20、0.50、1.0、2.0、4.0、6.0、10 μg/mL标准工作液,分别移取100 μL各标准工作液于15 mL塑料离心管中,各离心管中事先已加入2 mL去离子水,按1.2.1方法处理后测定。以峰面积为纵坐标,敌草快还原产物质量浓度为横坐标绘制标准曲线。经计算其回归方程为:Y=266.5X-3 326.98,r=0.999 7,表明在 0.20~10 μg/mL的范围内,峰面积与敌草快还原产物质量浓度之间线性关系良好。
2.4 回收率、精密度
采用本方法分别在菜籽油、大豆油、玉米油中添加敌草快进行加标回收实验,结果见表1。

表1 方法回收率、精密度实验结果(n=3)
从表1可以看出,方法加标回收率在90.6%~93.8%之间,相对标准偏差(RSD)为0.25%~3.01%。
2.5 仪器精密度
称取1 g (精确至0.001 g)大豆油空白样品,加入2.0 μg/mL标准工作液100 μL,按1.2.1方法处理,连续进样6次,考察仪器的精密度,结果见表2。
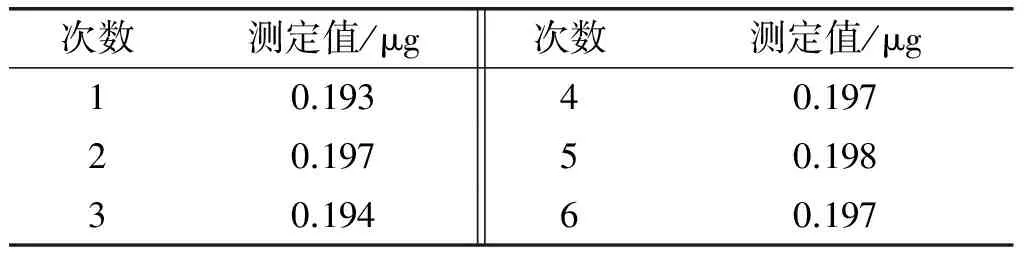
表2 仪器精密度实验结果
从表2可以看出,加标样品中敌草快含量的相对标准偏差为0.932%。说明仪器稳定性良好。
3 结 论
本文建立了液液萃取-气质联用法测定食用植物油中敌草快含量,样品前处理操作简便,方法重现性好。方法回收率在90.6%~93.8%之间,相对标准偏差为0.25%~3.01%。该方法快速、简便,适用于食用植物油中敌草快含量的快速检测。
[1] 朱震海, 宣栋梁. 固相萃取-HPLC 法测定水中敌草快, 百草枯[J]. 中国卫生检验杂志, 2011, 21(9): 2154-2156.
[2] 覃东立, 吴松, 郑敏, 等. 高效液相色谱法测定渔业水域中百草枯和敌草快[J]. 分析试验室, 2013, 32(2): 54-57.
[3] 陈静, 刘召金, 安保超, 等. 在线净化/固相萃取-高效液相色谱法测定饮用水和环境水体中的百草枯和敌草快[J]. 色谱, 2012, 30(10): 1068-1073.
[4] 曹慧, 陈小珍, 张东雷, 等. 液相色谱-串联质谱法快速测定粮谷中矮壮素和敌草快残留量[J]. 理化检验:化学分册, 2013, 49(4): 425-427.
[5] 王连珠, 李晓莲, 方恩华, 等. QuEChERS-液相色谱-串联质谱法测定棕榈原油中敌草快等 6 种农药残留[J]. 色谱, 2016, 34 (7):686-691.
[7] 李捷, 杨方, 卢声宇, 等. 超高效液相色谱-电喷雾串联质谱法测定茶叶中敌草快和百草枯残留[J]. 分析试验室, 2014, 33(5):537-541.
[8] 高利娜, 郑晓娇, 聂志伟, 等. 紫外分光光度法检测尿中敌草快[J]. 中国法医学杂志, 2014, 29 (6): 551-552.
[9] 朱雨田, 李锦才, 高素君,等. 近红外光谱技术在食用油快速检测领域中的研究进展[J].中国油脂, 2017, 42(7):140-143.
[10] DE ALMEIDA R M, YONAMINE M. Gas chromatographic-mass spectrometric method for the determination of the herbicides paraquat and diquat in plasma and urine samples[J]. J Chromatogr B, 2007, 853(1): 260-264.
[11] 高利娜, 宋洋, 祝娟, 等. 固相微萃取结合气质联用测定血浆中敌草快[J]. 中国法医学杂志, 2014,29 (5):427-430.
[12] 魏斌,朱臻怡, 张科,等. 气相色谱-质谱/负化学源法同时检测芝麻调和油中9种拟菊酯农药[J].中国油脂, 2016, 41(1):88-91.
[13] 杨广, 刘新, 鄢铮, 等. 4种拟除虫菊酯农药的GC-MS和GC-MS/MS检测[J]. 福建农林大学学报(自然科学版), 2003, 32(4):447-452.
[14] 杨君, 王建华, 刘靖靖, 等. 气相色谱-负化学离子源质谱法在食品安全分析中的应用[J]. 化学分析计量, 2012, 21(5):97-100.
[15] 陈劲星. 分散固相萃取-气相色谱-质谱法测定大豆油中102种农药残留量[J]. 福建分析测试, 2011, 20(1):23-28.
