掺杂三角形硼氮片的锯齿型石墨烯纳米带的磁电子学性质∗
2018-01-18张华林孙琳韩佳凝
张华林 孙琳 韩佳凝
(长沙理工大学物理与电子科学学院,长沙 410114)
(2017年7月26日收到;2017年9月17日收到修改稿)
1 引 言
自从2004年Novoselov等[1]成功制备单层石墨烯以来,石墨烯以其独特的性质引起了物理、化学、材料等学科研究人员的广泛关注.石墨烯纳米带是一种准一维碳纳米材料,根据边界结构不同,石墨烯纳米带分为锯齿型石墨烯纳米带(ZGNR)和扶手椅型石墨烯纳米带(AGNR).不考虑自旋时,ZGNR为金属;考虑自旋时,其铁磁(FM)态为金属,而反铁磁(AFM)态为半导体[2−5].对于石墨烯纳米带电子特性调制的研究已有很多报道,主要分为掺杂、吸附[6,7]、边缘修饰[8−10]和引入缺陷[11−13]等调控手段,其中掺杂对石墨烯的结构和电子性能的影响是一项非常值得关注的工作.在有关掺杂的研究中,有很多关于硼(B)和氮(N)原子掺杂的研究工作,而对于B和N掺杂,又设计有不同形式.有的在石墨烯纳米带中单独掺杂B或N原子[14,15],有的在一个较大超原胞中只掺杂一对B和N原子[16,17],有的成排掺杂B和N原子对[5,15],还有的将BN片掺杂到石墨烯中.Xu等[18]研究了BN片掺杂对石墨烯超晶格的电子与磁性的影响,发现不管BN片的形状是六边形还是三角形,石墨烯超晶格的带隙随着BN片的增大而增加.Manna和Pati[19]研究了嵌入BN片的二维石墨烯片的电子结构和磁性特征,研究表明嵌入BN的形状(含六边形和三角形)和大小对二维石墨烯片的电子结构有显著影响.Menezes和Capaz[20]研究了三角形h-BN分子团嵌入石墨烯超晶胞的电子结构和磁性.He等[21]提出了一种由锯齿型氮化硼纳米带和ZGNR构造的混合结构,并计算其能带结构及电子输运性质,结果表明混合结构的带隙可以调整,通过改变ZGNR的宽度可以实现从绝缘体到金属的转变,且不同自旋极化电流表现出不同的行为.Seol和Guo[22]研究了由BN纳米带约束的AGNRs的电子性质.
基于最近实验已实现在二维石墨烯中掺杂BN片的混合纳米结构[23],本文采用基于密度泛函理论(DFT)的第一性原理,探讨三角形BN片掺杂的ZGNR,在无磁(NM)态、FM态和AFM态三种情况下的磁电子学特性.这些研究结果对于发展基于石墨烯的纳米电子器件有重要意义.
2 计算模型与方法
BN片掺杂的ZGNR的几何结构如图1所示.图中蓝色虚线方框表示最小周期性重复单元,即计算选取的超原胞;白色、灰色、粉红色和蓝色原子分别为H,C,B和N原子;n表示完整ZGNR的带宽,即沿宽度方向的锯齿型碳原子链的排数.本文计算的模型选取带宽n=7的ZGNR作为代表.超原胞中的红色虚线三角形表示掺杂的位置,用Pi编号,其中i=1,2,3,4,5.图1中给出了P3位置掺杂的模型,P3位置的三角形内的所有C原子被BN原子取代,共6个B原子和7个N原子.为了消除悬挂键,所有ZGNR边缘用H原子饱和.

图1 (网刊彩色)三角形BN片掺杂的ZGNR的计算模型Fig.1.(color online)The geometric structure of ZGNR doped with triangular BN segment.
采用基于DFT的第一性原理的ATK(Atomistix ToolKit)软件包计算几何结构优化和电磁性质[24−26],目前它已被广泛用于纳米结构的研究中.模型结构在非磁性下进行优化.结构优化收敛的标准是作用在每个原子上的力小于0.02 eV/Å.为了求解Kohn-Sham方程,交换关联势采用广义梯度近似中的Perdew-Burke-Ernzerhof近似,赝势采用模守恒赝势,基函数组选用DZP(Doubleζ+Polarized).在简约布里渊区的k点抽样采用1×1×50.能量截取半径(mesh cut-off)采用150 Ry.X和Y方向超原胞的真空层设置为15 Å.
3 计算结果与讨论
3.1 NM态
ZGNR在NM态的能带结构和态密度(DOS)如图2所示,图2(a)–(f)分别表示未掺杂ZGNR和P1–P5位置掺杂的ZGNR(下同).图2中Z表示未掺杂ZGNR,Pi(i=1,2,3,4,5)表示掺杂的位置,BS表示能带结构,DOS表示态密度(下同),PDOS表示投影态密度,在此为投影到C原子的投影态密度.图2中纵轴为能量(E/eV),过纵轴零点的虚线表示费米能级(下同).各模型的超原胞未取代掺杂时共有42个碳原子,各掺杂模型被BN原子取代的C原子数为13个.定义掺杂浓度为被BN分子取代的C原子数与未取代掺杂时的C原子总数的比值,则各模型的掺杂浓度都为31.0%.由图2可见,掺杂后ZGNR的能带结构发生了一定的变形和移动.不论是否掺杂,ZGNR都有子能带穿越费米能级,且在紧靠费米能级的位置,穿越费米能级的子能带都有一段非常平缓.DOS与其相应的能带结构有很好的对应性,能带平缓处DOS出现峰值,能带陡峭处DOS较小,而没有能带分布处DOS为零.不论是否掺杂,ZGNR的DOS在费米能级处都出现一个较大的峰值.由此可见,不论是否掺杂,ZGNR都无带隙,都为金属.不论是否掺杂,ZGNR的PDOS图都与相应的DOS图都有很好的对应性,只是峰值大小略有差异,可见在能量为−1–1 eV范围内,ZGNR的DOS主要由C原子贡献.
为了更清楚地分析哪些原子对穿越费米能级的子能带起作用,图3绘出了各模型该子能带的电荷密度图,等值面取0.05|e|Å−3.由图3可见,ZGNR穿越费米能级的子能带主要由第1排和第7排C链中靠近边缘的C原子起作用,其次为第2排和第6排C链中靠近边缘的C原子.BN掺杂对掺杂区域的电荷密度具有明显的抑制作用.由此可见,由于掺杂位置的不同,电荷密度被抑制的区域不同,导致对各掺杂ZGNR的穿越费米能级的子能带起作用的原子不同,从而导致该子能带略有不同程度的移动和变形.

图2 NM态的能带结构和DOSFig.2.The band structure and density of states in the nonmagnetic state.

图3 (网刊彩色)部分子能带的电荷密度 (a)Z;(b)P1;(c)P2;(d)P3;(e)P4;(f)P5Fig.3.(color online)The charge density of partial subband:(a)Z;(b)P1;(c)P2;(d)P3;(e)P4;(f)P5.
3.2 FM态
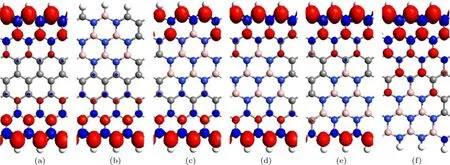
图4 (网刊彩色)自旋极化电荷密度等值面图 (a)Z;(b)P1;(c)P2;(d)P3;(e)P4;(f)P5Fig.4.(color online)The isosurface plots of spin polarization charge density in the ferromagnetic state:(a)Z;(b)P1;(c)P2;(d)P3;(e)P4;(f)P5.
ZGNR在FM态的自旋极化电荷密度等值面如图4所示.图4中红色和蓝色分别代表α和β自旋,等值面取0.01|e|−3(下同).自旋极化电荷密度Δρ=ρα−ρβ,其中ρα和ρβ分别表示α和β自旋的电荷密度.由图4可见,未掺杂的ZGNR和P2–P4位置掺杂的ZGNR的自旋极化电荷密度的共同特点是,上下两边缘的自旋方向相同,其磁性状态都表现为FM性.各掺杂的ZGNR中BN掺杂区域的自旋极化电荷密度均受到明显的抑制.这一点也可以从各模型的超原胞的总磁矩反映出来,未掺杂的ZGNR和P1–P5位置掺杂的ZGNR超原胞的总磁矩分别为1.51µB,0.60µB,1.00µB,1.01µB,1.00µB,0.95µB,µB为玻尔磁子.可见各掺杂的ZGNR的总磁矩明显小于未掺杂的ZGNR的总磁矩.对于P1和P5位置掺杂的ZGNR,由于都有BN原子位于纳米带的边缘,杂质所在边缘的自旋极化电荷密度受到抑制,这种情况其磁性状态也表现为FM性.由于B或N原子中的3个价电子与邻近的原子成键,B原子已没有剩余的未成键的价电子去贡献磁性,而N原子虽然还有2个剩余的未成键的价电子,但其自旋方向相反,无净磁矩,故BN掺杂对掺杂区域自旋极化电荷密度有明显的抑制作用[5].
ZGNR在FM态的能带结构和DOS如图5所示,图中蓝色实线和红色实线分别表示α和β自旋(下同).由图5可见,不论是否掺杂,ZGNR在费米能级附近的能带都发生了分裂,且α自旋的能带向下移动,β自旋的能带向上移动.结合图4和各模型的总磁矩为正,可以知道α自旋为多子态,而β自旋为少子态.正是因为α自旋是多子态,才导致费米能级附近α自旋的能带向下移动,以提供更多态让电子填充,而β自旋是少子态,导致β自旋的能带向上移动,减少可供电子填充的态.未掺杂ZGNR和P1位置掺杂的ZGNR的α和β自旋都有能带穿越费米能级,且在费米能级处的DOS都不为零,故都为自旋金属.P2–P4位置掺杂的ZGNR的α自旋都没有能带穿越费米能级,且在费米能级处的DOS都为零,而β自旋都有能带穿越费米能级,且在费米能级处的DOS都不为零,故都为自旋半金属.P2–P4位置掺杂的ZGNR的α自旋都具有直接带隙,导带底和价带顶都位于Γ点,带隙大小略有差异,分别为0.284,0.279,0.347 eV.自旋半金属对实现自旋过滤有重要意义.P5位置掺杂的ZGNR的α和β自旋都没有能带穿越费米能级,在费米能级处的DOS都为零,故为自旋半导体,且为双极化自旋半导体,其上旋和下旋带隙分别为0.341 eV和0.118 eV.双极化自旋半导体对于在改变栅极偏压正负时,实现双自旋过滤有重要意义.可见在ZGNR中掺杂三角形BN片,当处于FM态时,随着掺杂位置的改变,可以实现自旋金属-自旋半金属-自旋半导体的变化过程,且非常有意思的是只要掺杂区域不在边缘,掺杂的ZGNR就为自旋半金属.
3.3 AFM态
ZGNR在AFM态的自旋极化电荷密度等值面如图6所示.由图6可见,未掺杂的ZGNR和P2–P4位置掺杂的ZGNR的自旋极化电荷密度的共同特点是,上下两边缘的自旋方向相反,其磁性状态都表现为AFM性.掺杂的ZGNR中BN掺杂区域的自旋极化电荷密度均受到明显的抑制.P1和P5位置掺杂的ZGNR,由于掺杂区域都有BN原子位于纳米带的边缘,杂质所在边缘的自旋极化电荷密度受到抑制,这种情况其磁性状态依然表现为FM性,即P1和P5位置掺杂的ZGNR没有AFM态.
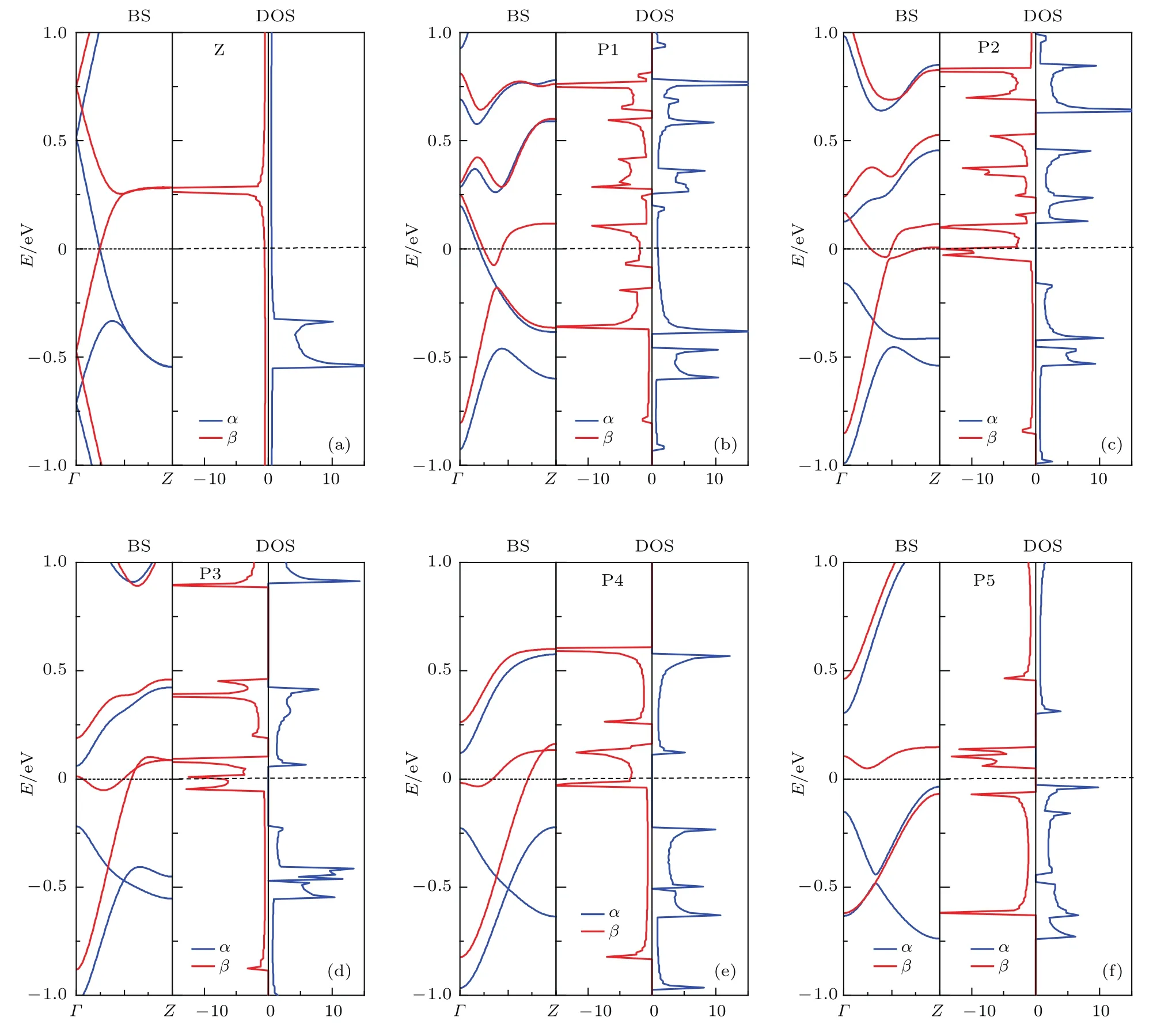
图5 (网刊彩色)FM态的能带结构和DOSFig.5.(color online)The band structure and density of states in the ferromagnetic state.
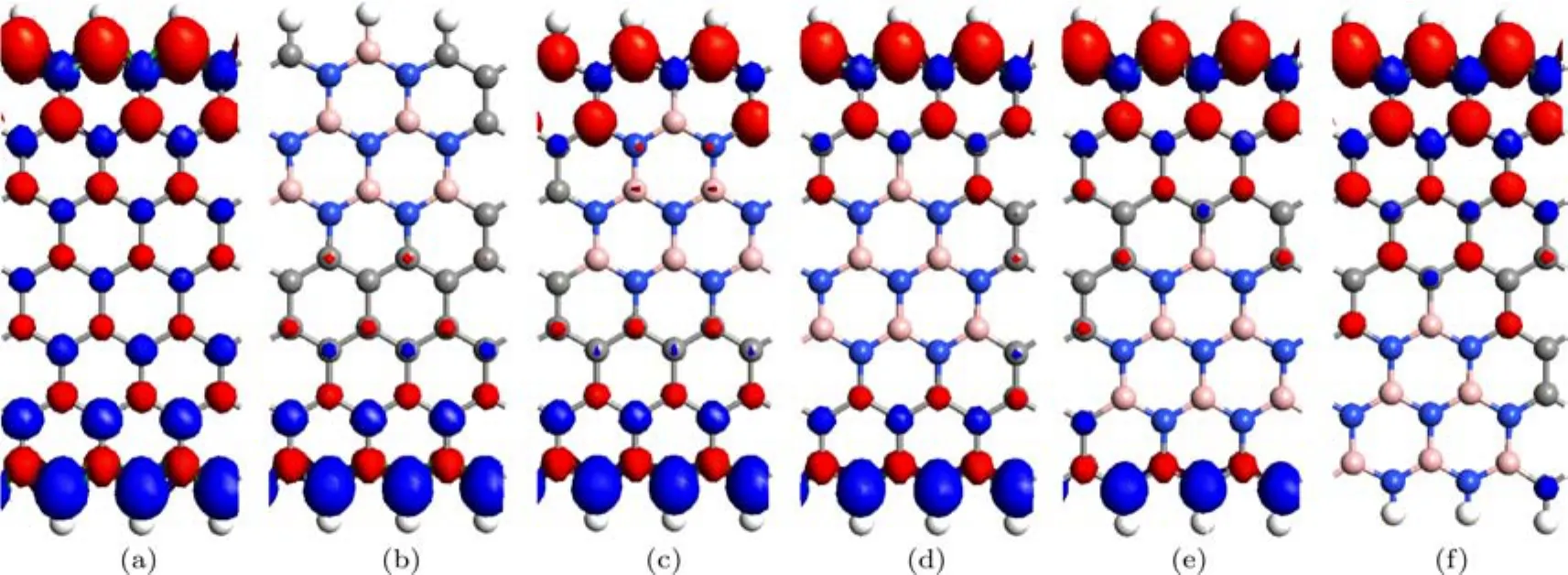
图6 (网刊彩色)自旋极化电荷密度等值面图 (a)Z;(b)P1;(c)P2;(d)P3;(e)P4;(f)P5Fig.6.(color online)The isosurface plots of spin polarization charge density in the antiferromagnetic state:(a)Z;(b)P1;(c)P2;(d)P3;(e)P4;(f)P5.

图7 (网刊彩色)AFM态的能带结构和DOSFig.7.(color online)The band structure and density of states in the antiferromagnetic state.
ZGNR在AFM态的能带结构和DOS如图7所示.由图7可见,未掺杂ZGNR在费米能级附近的能带是简并的,没有能带穿越费米能级,且在费米能级处的DOS为零,故为半导体,其α和β自旋的带隙大小都为0.518 eV.未掺杂ZGNR的α和β自旋极化电荷密度关于对称轴成对称分布,总磁矩为零,α和β自旋电子是均衡的,故导致其能带简并.但掺杂的ZGNR在费米能级附近的能带都发生了分裂,这是因为BN掺杂区域的自旋极化电荷密度受到抑制,导致α和β自旋电子不再均衡.P2–P4位置掺杂的ZGNR的α和β自旋都有能带穿越费米能级,且在费米能级处的DOS都不为零,故都为自旋金属.结合图5和图7可见,P5位置掺杂的ZGNR在FM态和AFM态的能带结构是相同的,因为两种自旋极化电荷密度是相同的,总磁矩都是0.95µB,都是α自旋为多子态;而P1位置掺杂的ZGNR在FM态和AFM态的能带结构形式相同,只是α和β自旋正好交换了,这是因为在FM态时α自旋为多子态,AFM态的总磁矩为−0.60µB,其β自旋为多子态,由对称性可知,其AFM态实质上还是FM态.这进一步说明了P1和P5位置掺杂的ZGNR没有AFM态.
3.4 结构稳定性分析
为了研究取代掺杂结构的稳定性,分别取各位置掺杂的ZGNR的超原胞,计算结合能和不同磁性态的总能差.各位置掺杂的ZGNR的总能差(ΔE/meV)和结合能如图8所示.定义FM态与AFM态的总能差ΔE1=EFM−EAFM,NM态与AFM态的总能差ΔE2=ENM−EAFM,EFM,EAFM和ENM分别为各超原胞在FM态、AFM态和NM态的总能.定义结合能

式中,Edop为掺杂ZGNR的超原胞的总能量;EC,EB,EN,EH分别为单个C,B,N,H原子的能量;nC,nB,nN,nH分别为超原胞内C,B,N,H原子的数量[27].由图8可见,对于各位置掺杂的ZGNR都有ΔE2>0,即ENM>EAFM;对于P2–P4位置掺杂的ZGNR有ΔE1>0,即EFM>EAFM;而对于P1和P5位置掺杂的ZGNR有ΔE1=0,即EFM=EAFM,表明P2–P4位置掺杂的ZGNR的基态是AFM态,P1和P5位置掺杂的ZGNR没有AFM态,其基态为FM态.发现P1–P5位置掺杂的ZGNR的结合能都为负值,且结合能的大小都与参考文献[27]中的数值(−8.13 eV)非常接近,说明掺杂的ZGNR的结构是稳定的.随着掺杂位置由P1向P5移动,结合能逐渐减小,P5位置掺杂的ZGNR的结合能最小,说明在P5位置掺杂的ZGNR的结构最稳定.
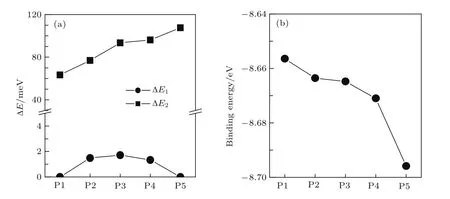
图8 各位置掺杂的ZGNR的(a)总能差和(b)结合能Fig.8.(a)The total energy differences and(b)the binding energies for the ZGNRs doped with triangular BN segments at different positions.
3.5 尺度效应
为了研究在不同宽度ZGNR中相同位置掺杂相同大小的BN片的磁电子学特性,在此建立两个与P2模型类似的模型,ZGNR的带宽分别为n=6和8,即保持BN片的大小和位置不变,将n=7的ZGNR模型的下边缘分别减少和增加一排碳链,这两个模型分别用Z6和Z8表示.Z6和Z8模型的掺杂浓度分别为36.1%和27.1%,即随着ZGNR带宽的增大,掺杂浓度减小,计算参数同上.Z6和Z8模型在NM,FM和AFM态的能带结构如图9所示.由图9可见,在NM态,Z6和Z8模型都有能带穿越费米能级,都为金属.结合图2(c)可知,当处于NM态时,在P2位置掺杂相同大小的BN片,改变ZGNR的宽度,其属性不变,都为金属.在FM态,Z6和Z8模型的α自旋都没有能带穿越费米能级,而β自旋都有能带穿越费米能级,故都为自旋半金属.结合图5(c)可知,当处于FM态时,在P2位置掺杂相同大小的BN片,改变ZGNR的宽度,其属性不变,都为自旋半金属.带宽n=6,7,8的掺杂ZGNR的上旋能带都有直接带隙,其带隙大小分别为0.373,0.284,0.234 eV,可见在所研究的带宽范围内,随着ZGNR带宽的增大,上旋带隙逐渐减小.在AFM态,Z6和Z8模型的α自旋和β自旋都有能带穿越费米能级,故都为自旋金属.结合图7(c)可知,当处于AFM态时,在P2位置掺杂相同大小BN片,改变ZGNR的宽度,其属性不变,都为自旋金属.可见在所研究的带宽范围内,保持BN掺杂区域的大小和位置不变,改变ZGNR的带宽,三种磁性态下各自的属性保持不变.

图9 (网刊彩色)不同宽度掺杂ZGNR的能带结构 (a)NM;(b)FM;(c)AFMFig.9.(color online)The band structures of doping ZGNRs with different widths:(a)NM;(b)FM;(c)AFM.
4 结 论
利用基于DFT的第一性原理研究了三角形BN片掺杂对ZGNR的磁电子学性质的影响.研究表明:当处于NM态时,各位置掺杂的ZGNR都为金属.BN掺杂对ZGNR的自旋极化电荷密度具有明显的抑制作用,导致考虑自旋时,不同位置掺杂的ZGNR呈现不同的属性;当处于FM态时,随着杂质在ZGNR中由P1位置向P5位置移动,依次可以实现自旋金属-自旋半金属-自旋半导体的变化过程,且只要掺杂区域不在ZGNR的边缘,掺杂的ZGNR就为自旋半金属;当处于AFM态时,P2–P4位置掺杂的ZGNR都为自旋金属.P1和P5位置掺杂的ZGNR没有AFM态.P2–P4位置掺杂的ZGNR的基态是AFM态,P1和P5位置掺杂的ZGNR的基态为FM态.P1–P5位置掺杂的ZGNR的结合能都为负值,且随着掺杂位置由P1向P5移动,结合能逐渐减小,P5位置掺杂的ZGNR的结合能最小,说明掺杂的ZGNR的结构是稳定的,且在P5位置掺杂最稳定.该研究对于发展基于石墨烯的纳米电子器件很有意义.
[1]Novoselov K S,Geim A K,Morozov S V,Jiang D,Zhang Y,Dubonos S V,Grigorieva I V,Firsov A A 2004Science306 666
[2]Barone V,Hod O,Scuseria G E 2006Nano Lett.6 2748
[3]Yang L,Park C H,Son Y W,Cohen M L,Louie S G 2007Phys.Rev.Lett.99 186801
[4]Hod O,Barone V,Scuseria G E 2008Phys.Rev.B77 035411
[5]Wang D,Zhang Z H,Deng X Q,Fan Z Q 2013Acta Phys.Sin.62 207101(in Chinese)[王鼎,张振华,邓小清,范志强2013物理学报62 207101]
[6]Farzaneh S 2015J.Phys.Chem.C119 12681
[7]Wang Q H,Shih C J,Paulus G L C,Strano M S 2013J.Am.Chem.Soc.135 18866
[8]Kan Z,Nelson C,Khatun M 2014J.Appl.Phys.115 153704
[9]Zhu Z,Zhang Z H,Wang D,Deng X Q,Fan Z Q,Tang G P 2015J.Mater.Chem.C3 9657
[10]Yu Z L,Wang D,Zhu Z,Zhang Z H 2015Phys.Chem.Chem.Phys.17 24020
[11]Zhang W X,He C,Li T,Gong S B 2015RSC Adv.5 33407
[12]Tang G P,Zhang Z H,Deng X Q,Fan Z Q,Zhu H L 2015Phys.Chem.Chem.Phys.17 638
[13]Zhang H L,Sun L,Wang D 2016Acta Phys.Sin.65 016101(in Chinese)[张华林,孙琳,王鼎 2016物理学报65 016101]
[14]Zhao S Q,Lü Y,Lü W G,Liang W J,Wang E G 2014Chin.Phys.B23 067305
[15]Liu J,Zhang Z H,Deng X Q,Fan Z Q,Tang G P 2015Org.Electron.18 135
[16]Xiao J,Yang Z X,Xie W T,Xiao L X,Xu H,Ouyang F P 2012Chin.Phys.B21 027102
[17]Liu Z M,Zhu Y,Yang Z Q 2011J.Chem.Phys.134 074708
[18]Xu B,Lu Y H,Feng Y P,Lin J Y 2010J.Appl.Phys.108 073711
[19]Manna A K,Pati S K 2011J.Phys.Chem.C115 10842
[20]Menezes M G,Capaz R B 2012Phys.Rev.B86 195413
[21]He J,Chen K Q,Fan Z Q,Tang L M,Hu W P 2010Appl.Phys.Lett.97 193305
[22]Seol G,Guo J 2011Appl.Phys.Lett.98 143107
[23]Ci L J,Song L,Jin C H,Jariwala D,Wu D X,Li Y J,Srivastava A,Wang Z F,Storr K,Balicas L,Liu F,Ajayan P M 2010Nat.Mater.9 430
[24]Hu R,Fan Z Q,Zhang Z H 2017Acta Phys.Sin.66 138501(in Chinese)[胡锐,范志强,张振华2017物理学报66 138501]
[25]Zhang Z,Zhang J,Kwong G,Li J,Fan Z,Deng X,Tang G 2013Sci.Rep.3 2575
[26]Zhang Z H,Guo C,Kwong D J,Li J,Deng X Q,Fan Z Q 2013Adv.Funct.Mater.23 2765
[27]Kan M,Zhou J,Sun Q,Wang Q,Kawazoe Y,Jena P 2012Phys.Rev.B85 155450
