脑小血管病和脑微循环研究进展
2018-01-12刘品一黄丽丽徐运
刘品一,黄丽丽,徐运
1 概述
脑小血管病(cerebral small vessel disease,CSVD)是由大脑的小动脉(直径40~200 μm)、穿支动脉、毛细血管及小静脉等小血管的各种结构或功能性的病变所导致的临床、认知、影像学及病理表现的综合征[1-3]。CSVD不但占卒中的20%~30%,更显著地提高了未来卒中的发病率[4]。近年来,CSVD因其发病的隐匿性及临床表现的多样性越来越引起学者的关注,2008年世界卒中日主题就是“小卒中,大问题”[5]。有研究发现,在一个平均年龄为(62±9)岁的群体研究中,1/10的参与者经磁共振成像(magnetic resonance imaging,MRI)后发现存在亚临床脑梗死[5-6]。许多研究也发现,随着年龄的增长,隐匿性卒中(又称静息性卒中)的发病率随之增加[6],导致许多老年人出现急性卒中、认知功能损害、记忆力下降、构音障碍、抑郁、步态不稳、泌尿系统障碍、痴呆等症状[2,7-8],极大地增加了社会经济负担。但是CSVD的病因至今不明,临床上病理学检查有所发现时常常已是疾病的终末期,而CSVD的微血管损伤往往先于脑实质的损伤,因此,明确CSVD脑微循环相关性的病理生理机制并加以干预具有重要意义。
CSVD在临床上有多种分类方法。由于活体上难以直接观察CSVD的发生及变化,因此,临床上多以神经影像学特征作为CSVD的观察指标,可分为腔隙性梗死(lacunar infarction,LI)、脑白质高信号(white matter hyperintensities,WMH)、脑微出血(cerebral microbleed,CMB)、血管周围间隙扩大(enlarged perivascular spaces,EPVS)和脑萎缩(cerebral atrophy)[2]。按CSVD的动脉病理学分类见表1[1]。散发型和遗传性CSVD则是按照其遗传特征进行的分类。
2 脑微循环
脑微循环即脑的微动脉和微静脉之间的血液循环,其基本功能是完成血液和组织之间的物质交换。
脑部微小动脉有两种血管来源,一种是蛛网膜下隙的软脑膜动脉分支的终末动脉支,另一种是直接来源于大血管的供应脑深部的穿支动脉支。这两种动脉系统相向而行,分别穿透脑皮质层和深部髓质,在皮质下白质的深处汇合,形成分水岭区[1]。此处的小动脉均为终末支,无吻合支,且血管密度低,故极易发生局部的循环缺血和低灌注[3],急性缺血易发生腔隙性梗死或CMB,慢性则可导致脑白质病变。终末微动脉的血流形成毛细血管网,经毛细血管后微静脉汇入硬膜下静脉窦[1]。
脑小动脉有其独有的循环特点[3]:①大脑几乎没有能量储备功能,故脑血流量对其影响大。虽然人脑只占了体重的2%,但消耗了心排血量的20%来维持正常脑功能[9]。但是人脑缺乏能量储备功能,故必须依靠持续性的血流灌注来提供所需的葡萄糖、氧气及其他营养物质[10]。静止时脑血流量(cerebral blood flow,CBF)的决定因素有灌注压、自动调节机制、血管对二氧化碳分压的反应性。脑微循环中多种细胞,包括星形胶质细胞、血管平滑肌细胞、内皮细胞和周细胞等在维持组织灌注压和血流动力学稳定方面发挥重要作用[7],使微循环的血流量与组织代谢水平相一致。②日常活动中脑动脉压的波动范围大,大脑血管的自动调节机制可在一定的动脉压范围内保持脑血流的相对恒定。③脑微循环有其独特的结构即血脑屏障(blood-brain barrier,BBB),BBB由内皮细胞、紧密连接蛋白及基底膜构成。在毛细血管水平,BBB的完整性与周细胞、血管周星形胶质细胞等也密切相关。
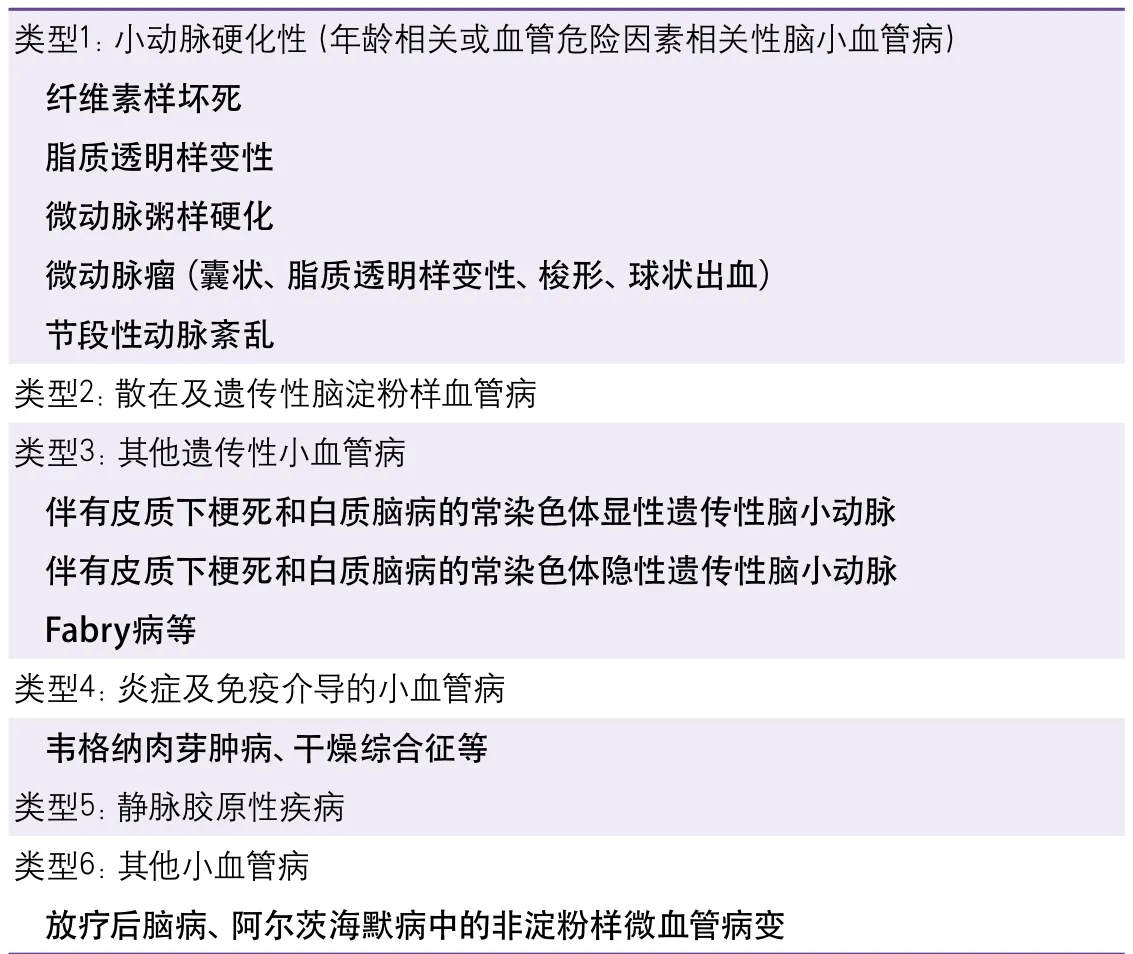
表1 脑小血管病的动脉病因学分类
3 脑微循环相关性CSVD的病理机制
3.1 小动脉硬化与栓塞 Fisher[11-13]最早提出CVSD的发病与小动脉硬化与栓塞有关,他发现CSVD最常见的病理学改变为小动脉(直径为40~200 μm)的弥漫性病变,表现为小动脉硬化、脂质透明样变及纤维素样坏死。另外,在Fisher进行病理学检查的10例腔隙性梗死患者中,有6例梗死可归因于穿支动脉的粥样斑块,2例归因于脂质透明样变,2例归因于栓塞[13]。尽管如此,有一系列的队列研究和meta分析表明,只有10%~15%的腔隙性梗死和少数的WMH是由栓子引起[14-16]。此外,将栓子注入实验模型的颈动脉后发现,只有极少数(<6%)的栓子进入穿支动脉[17-18]。与非腔隙性缺血性卒中相比,腔隙性卒中与常见栓子来源(如心房颤动或同侧近端颈动脉狭窄)的相关性更弱[14-15]。
高脂血症、高血压、吸烟、糖尿病等危险因素被认为与小动脉硬化发病密切相关。小动脉硬化的病理特征为中膜平滑肌细胞的丢失,纤维、玻璃样物质沉积,管壁增厚,管腔狭窄,并可继发血栓形成加重狭窄甚至闭塞。血管管腔狭窄使脑白质处于慢性低灌注状态,最终导致少突胶质细胞死亡,即有髓神经纤维退化[1]。这一缺血机制已通过动物实验得以证实[19-20]。这种脑白质损伤被认为是一种不完全性梗死或选择性坏死[21]。此外,Fisher[11-12]提出急性血管闭塞可导致局部急性缺血和完全性组织坏死,即腔隙性梗死。但是,这一假说尚未得到证实[22-23]。
另外,小动脉硬化时,血管壁中膜的平滑肌细胞丢失,可导致血管弹性降低[24-25]。因此,脑小血管的舒缩功能受到影响,即自动调节机制受损,进而抑制了栓子清除能力,使脑血流量失控,最终引发栓塞[26-27]。类似的,由于血管弹性降低,小动脉搏动减弱,而细胞间液引流需要依靠邻近小动脉搏动所产生的挤压作用,因此细胞间液引流出现障碍,细胞间液对脑细胞具有毒性作用,可导致白质疏松及脱髓鞘病变,在影像学上表现为WMH及血管周围间隙扩大[28-31]。
3.2 内皮细胞功能障碍与BBB破坏 虽然小动脉硬化、栓塞可引起CSVD相关的病理生理改变,但这通常发生在CSVD的后期,无法反映CSVD早期的病理生理变化[2]。一些学者认为内皮细胞功能障碍及BBB破坏是CSVD早期脑损害的潜在机制[32]。血浆内的有害成分可通过受损的内皮及BBB溢出管腔,进而导致血管壁肌细胞的损伤以及周围脑实质的弥漫性损害[33]。钆增强MRI显示腔隙性卒中患者的BBB通透性大于皮质卒中患者,WMH处通透性大于脑白质正常处[34-35]。尽管磁共振(magnetic resonance,MR)监测到的钆信号很低,但这依然能够表明随着年龄增长及血管周围间隙数量增多,BBB通透性增加[34]。另外,通过对脑组织的组织病理学检查可以发现CSVD的严重程度与血管外的血浆蛋白(如纤维蛋白原、白蛋白、免疫球蛋白G)有关[36-37],但也有部分研究并未发现其中的关系[38-39]。
高血压是CSVD最常见的危险因素,而内皮细胞功能障碍是其中的一个重要环节[40]。自发性高血压大鼠(spontaneously hypertensive rats,SHRs)和易卒中自发性高血压大鼠(stroke-prone SHRs,SHR-SPs)都表现出内皮细胞功能障碍,如磷酸化内皮一氧化氮(nitric oxide,NO)合成酶表达下调及BBB破坏[41]。SHRs与SHR-SPs的高血压水平相似,但脑白质损伤程度不同[42]。在高血压发病前,SHR-SPs就已经出现内皮、基质蛋白、胶质细胞及髓鞘受损,这些都是CSVD发病的基础,而高血压会使损伤进一步加重[43-44]。另外,一项关于CSVD的MRI研究表明,不仅高血压可以作为轻度卒中患者WMH损伤的预测指标,食盐量亦独立于高血压与WMH有关[40]。众所周知,高血压可分为盐抵抗性高血压和盐敏感性高血压,后者更易出现内皮细胞功能障碍,通常表现为内皮依赖性血管舒张受限[45]。此外,食盐量与炎症反应有关,SHR-SPs摄盐后脑组织与血清内的内皮细胞表达的炎症指标,即内皮细胞选择素显著上升,但SHRs并未发生改变[46]。高血压、高盐饮食等危险因素可直接或间接地影响内皮功能,继而破坏BBB,最终导致血管周围间隙扩大,CMB及脑白质改变[33]。
炎症与CSVD之间也存在着关联性[47-48]。炎症反应可通过激活的单核巨噬细胞分泌的新蝶呤和细胞因子作用于内皮细胞,进而破坏BBB[47,49-50]。而感染则是引起这种炎症反应的因素之一。Nakano等[51]发现变形链球菌的一个特定菌株是SHR-SPs及光化学诱导大脑中动脉闭塞小鼠发生脑出血的潜在危险因素。导致脑出血的变形链球菌菌株可在其细胞表面表达胶原结合蛋白,进而紧密黏附于受损血管表面暴露的胶原纤维,最终抑制血小板的激活,导致脑出血[51]。一项基于人群的研究表明胶原结合蛋白阳性变形链球菌与CMB的关联性很强(OR=14.4)[52]。两项研究结果相符。另外,Pussinen等[53]发现牙龈卟啉单胞菌与缺血性卒中相关。牙龈卟啉单胞菌黏附并感染内皮细胞,这样不仅可以使内皮黏附分子表达上调,促进单核巨噬细胞浸润,还可以产生牙龈素(半胱氨酸蛋白酶),从而激活血小板上的蛋白酶激活受体-1和-4,诱导血小板的聚集[54-55]。总之,口腔内的细菌不仅可以通过调节血小板的聚集,还可以通过炎症反应破坏BBB来引起CSVD相关的病理生理改变。这一机制被称为“脑-口腔轴”[56]。这可能是因为口腔与脑位置邻近,口腔内的细菌可以通过血液循环到达脑的相应位置并导致疾病的发生[56]。
心血管危险因素可以通过大动脉与小动脉间的相互作用同时损伤大、小动脉[57-58]。但大动脉病变究竟能否导致CSVD的发生还有待进一步研究[56]。利用微弹簧圈造成小鼠双侧颈总动脉狭窄,由此建立慢性低灌注及认知障碍的动物模型[59]。这一模型可以出现以BBB破坏为特征的脑白质损伤[60]。手术造成双侧颈总动脉狭窄2 h后,血流动力学发生改变,内皮细胞紧密连接开放,而基质金属蛋白酶-2(matrix metalloproteinase-2,MMP-2)抑制剂可以抑制这一过程[61-62]。MMP-2是一种细胞外基质降解酶,可以降解紧密连接蛋白ZO-1、claudin-5及封闭蛋白,从而导致BBB破坏[63]。MMP-2在SHR-SPs体内的表达水平上调,而且在人体内与WMH有关[64-65]。这些数据表明,大动脉狭窄所致低灌注状态可通过MMP-2在毛细血管水平损伤内皮细胞[56]。BBB功能障碍可导致蛋白质(如蛋白酶及免疫球蛋白)及液体渗漏,最终导致与脑白质损伤相关的病理改变,如脱髓鞘和胶质增生[32,66]。尽管如此,并没有证据能够直接证明颈动脉狭窄与CSVD相关。
上文已介绍BBB由内皮细胞、周细胞、星形胶质细胞、少突胶质细胞等成分构成。它们对维持正常的BBB起着十分重要的作用。但是周细胞、星形胶质细胞及少突胶质细胞与CSVD发病的关系尚未明晰。一项关于伴有皮质下梗死和白质脑病的常染色体显性遗传性脑小动脉(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CADASIL)的研究发现致病基因NOTCH3在周细胞中特异性表达,这提示周细胞与内皮细胞的相互作用可能与CSVD的发病相关[67]。一些研究报道血小板源性生长因子受体-β(platelet-derived growth factor receptor-β,PDGFR-β)阳性周细胞的数量在CADASIL患者的脑白质中增多[68-69]。因为周细胞可控制神经血管功能,其丢失可能预示着神经退行性病变,PDGFR-β阳性周细胞数量的增多可能与慢性低灌注状态下的代偿机制有关[70]。另外,最近有研究表明,星形胶质细胞分泌的脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)能够在脑白质缺血损伤期间促进少突胶质前体细胞的成熟[71]。胶质细胞与内皮细胞的相互作用能够在生理及病理状态下影响BBB[72]。少突胶质前体细胞与周细胞能够通过基膜相互黏附、相互作用,从而促进其增殖与分化[73]。以上的数据表明BBB的各个成分并非独立存在,它们可以相互作用,相互影响。如果各个成分
间的相互关系受到破坏,则可能会导致BBB功能障碍,从而诱发CSVD,而这其中的机制还有待进一步研究。
3.3 脑血流低灌注 Nakane等[74]的研究发现,静息性脑梗死患者的脑血流量较正常对照组明显降低,但较症状性脑梗死患者高。一项定量磁共振灌注成像研究也发现,在脑白质损害患者中,其白质病变区域的脑血流量较白质正常区域显著降低[75]。这些结果都表明脑微循环低灌注在CSVD的发病机制中有重要的意义。脑低灌注的原因有血管痉挛,小动脉硬化时平滑肌细胞的丢失、管壁的增厚所导致的自动调节机制的损害等[76],而血流低灌注通过多种机制导致CSVD病变:①通过血管内皮的损害导致BBB的功能障碍:低灌注导致MMP-2的增加,降解紧密连接蛋白[56,61],导致紧密连接的开放,BBB的结构破坏、渗透性增加。②一氧化氮合酶的乙酰化:Hattori等[62]发现沉默信息调节因子2相关酶I(SIRT1)-一氧化氮合酶(NOS)-NO轴参与调节脑内生理性能量的平衡,SIRT1过表达模型中其脱乙酰化作用,激活了NOS,合成NO调节脑血流量,使低灌注状态下仍能保持脑血流量的稳定。也为CSVD的治疗提供了新策略。③神经胶质细胞的激活:Shibata等[77]发现在双侧颈动脉狭窄(bilateral carotid artery stenosis,BCAS)的鼠模型中,脑白质病变区激活的小胶质细胞和星形胶质细胞有明显的增殖。Holland等[60]同样发现BCAS模型鼠在第6个月时胶质纤维酸性蛋白阳性的星形胶质细胞的数量有显著的增加。神经胶质细胞的激活常伴随星形胶质细胞终足的水通道蛋白AQP4的重新分布:血管内AQP4的数量有明显的下降而血管外实质内的AQP4则显著升高。④少突胶质细胞的凋亡:在人脑的发育过程中,室周的脑白质最易在妊娠的23周到32周因低氧缺血而发生损害,即所谓的脑室周围白质疏松(periventricular leukomalacia,PVL),其主要的血管因素是此阶段脑供血机制的不成熟和脑血流自动调节机制的易损性,但其细胞层面的机制犹未可知。由于PVL导致了慢性髓鞘形成的紊乱,故Back等[78]考虑少突胶质细胞为缺血损伤的靶细胞。进一步的实验研究发现,少突胶质细胞系对低氧缺血的易损性为成熟依赖性,其中晚期的少突胶质细胞祖细胞为导致PVL的主要高危阶段,而未成熟少突胶质细胞则对低氧缺血有一定的抵抗力,这也可以解释为何妊娠30周后PVL的发病率明显下降,因为此阶段正好是晚期少突胶质祖细胞发生分化而白质中未成熟少突胶质细胞增加至3倍的时期。晚期少突胶质祖细胞的凋亡主要与远端细胞色素C的释放和半胱氨酸天冬氨酸蛋白酶-3(caspase-3)的活化有关[78]。⑤氧化应激导致血管功能障碍[4]。⑥缺血继发的炎症反应,进而导致血管内皮的损伤或如上文所述的因炎症而引起CSVD。⑦缺血引发淀粉样脑血管病。
3.4 淀粉样蛋白沉积 脑淀粉样血管病是一种以皮质及脑膜间隙动脉、小动脉、静脉、毛细血管壁的中膜或外膜发生淀粉样蛋白沉积为特征的疾病[1]。它与CSVD有一定关联,可表现为CMB及WMH[79]。在疾病早期,淀粉样蛋白沉积可导致血管壁增厚,管腔狭窄,进展期管壁变薄,管腔扩张,严重时可出现小动脉瘤[80-82]。因此,脑淀粉样血管病可影响血流灌注,从而造成皮质下脑白质损伤以及组织结构的改变[83-86]。另外,对于脑淀粉样血管病患者而言,血管壁越厚,CMB风险越高[87]。这其中的机制还尚未明晰。但是,Kövari等[88]发现CMB常发生于额叶及顶叶,而脑淀粉样血管病常发生于枕叶;CMB常累及深部动脉及小动脉,而脑淀粉样血管病常侵犯皮质,主要是软脑膜动脉及其浅表皮质分支。这些证据对脑淀粉样血管病与CMB的相关性提出了质疑。由此可见,淀粉样蛋白沉积与CSVD的关系还有待进一步研究。
3.5 静脉胶原性疾病 静脉胶原性疾病是指静脉或小静脉壁的胶原成分增多,导致管壁增厚、管腔狭窄,甚至闭塞[1,89]。高血压及年龄增长可能与其发病相关[89-90]。Moody等[91]发现静脉胶原性疾病常发生于侧脑室周围,且与脑白质疏松有关。另外,一项与脑白质疏松部位深髓静脉相关的影像学研究表明大脑深部静脉缺血与WMH有关[92]。脑白质疏松可偶尔出现于侧脑室周围以外的区域,如皮质附近。在这种情况下,相比于侧脑室周围,静脉胶原性疾病更多地出现于脑白质疏松部位[89]。这也支持了静脉胶原性疾病与脑白质疏松有关的结论。静脉狭窄可导致静脉压增高,从而引起慢性缺血、静脉回流障碍、细胞间液重吸收障碍及脑水肿[91]。细胞间液体可对细胞及髓鞘产生毒性作用,导致退行性病变、脱髓鞘及胶质增生,在影像学上表现为WMH[91]。
4 脑微循环临床检查的方法
如上文所述,CSVD可有不同的影像学表现。影像学检查,尤其是MRI是诊断CSVD最重要的检查手段之一。相比MRI,计算机断层扫描(computed tomography,CT)仅在鉴别诊断方面有一定的临床意义。但是,常规的影像学检查无法提供与CSVD时脑微循环改变相关的信息。因此,我们需要特殊的影像学及实验室检查手段来观察脑微循环的改变。
4.1 脑血流动力学检查 脑血流量降低、脑血管反应性及其自动调节功能受损与CSVD发病相关[74-76]。MR灌注成像技术在脑血流动力学的检测中有着广泛的应用。MR灌注成像技术可分为内源性示踪技术和外源性示踪技术。外源性示踪技术是指将示踪剂(如钆)以静脉注射的方式注入体内,通过观察示踪剂通过组织信号变化的情况,检测不同的血流动力学指标,如脑血容量、脑血流速度及平均通过时间[93]。内源性示踪技术,即动脉自旋标记技术(arterial spin labeling,ASL),是一种以动脉血中的质子作为示踪剂,从而获得脑血流量的定性或定量图像的无创性技术[93]。与MR灌注成像技术类似,正电子发射型计算机断层成像技术(positron emission tomography,PET)利用正电子核素标记的示踪剂(如15O-H2O、18F-FDG)进行脑成像,进而评估脑血流量[93-94]。PET具有空间分辨率高、灵敏度高的特点,因此被认为是检测脑血流量的“金标准”,但其高昂的检查费用限制了它在临床的应用[93,95]。另外,经颅多普勒超声在评价脑血管反应性及其自动调节功能方面应用较广[93]。
4.2 内皮细胞及BBB功能检查 内皮细胞功能障碍可导致血管壁通透性增高,BBB破坏,血管内的血浆蛋白因而可以渗出血管壁,然后进入脑脊液中。所以,检测脑脊液/人血白蛋白比值是评估内皮细胞功能及BBB完整性最为经典的检查手段[93]。与健康人群相比,CSVD及出现脑白质不同程度改变的患者的脑脊液/人血白蛋白比值上高[96-98]。除此以外,一种名为二乙烯三胺五乙酸钆(gadolinium diethylene triamine pentaacetic acid,GD-DTPA)的造影剂也被用于评估BBB的通透性[93]。一项有关血管性认知障碍的研究表明,WMH区域与造影剂高通透区域之间有重叠,这提示血管源性水肿可导致继发性缺氧,从而引起脑白质病变[99]。在CSVD的发病过程中,一些与内皮细胞损伤及BBB破坏的生物学标记物表达上调,如细胞间黏附分子-1(intercellular cell adhesion molecule-1,ICAM-1)、组织金属蛋白酶抑制剂-1(tissue inhibitors of metalloproteinases,TIMP-1)、基质金属蛋白酶-9(matrix metalloproteinase-9,MMP-9)等[100-101]。但是这些指标尚未纳入临床检查的范围,更具特异性及临床意义的标记物还有待发现。
5 小结与展望
目前人们对CSVD的了解主要来源于影像学检查,但影像学改变并不能反映CSVD早期的病理生理改变。近年来,微循环障碍,特别是内皮细胞及BBB功能障碍被认为在脑小血管的发病过程中发挥着至关重要的作用。各种血管性危险因素、炎症、感染、大动脉病变均可损伤内皮细胞功能,进而影响BBB的完整性,从而导致CSVD的发生及相应的影像学改变[56]。但其中具体的分子机制还有待进一步阐明。另外,在静脉胶原性疾病及脑淀粉样血管病中,血管壁胶原成分增多、淀粉样蛋白沉积则是引起脑组织损伤的主要因素[1]。针对这些危险因素和已知发病机制中的重要环节,我们可采取相应的预防措施(如控制血压、低盐饮食、预防感染)并发展具有特异性的靶向治疗方法。有研究发现,MMP抑制剂可以抑制细胞外基质的降解,减轻血管内皮细胞的炎症反应,从而使BBB的完整性得到恢复[102]。目前脑微循环的临床检查不多,主要是通过影像学技术检测脑血流动力学的改变,其中ASL等无创性检查将在未来受到更多的关注。此外,更具特异性的生物标记物将有助于医学科研工作者评估内皮细胞及BBB的功能并在疾病早期及时采取干预措施。
1 Pantoni L.Cerebral small vessel disease:from pathogenesis and clinical characteristics to therapeutic challenges[J].Lancet Neurol,2010,9:689-701.
2 Wardlaw JM,Smith C,Dichgans M.Mechanisms of sporadic cerebral small vessel disease:insights from neuroimaging[J].Lancet Neurol,2013,12:483-497.
3 Joutel A,Faraci FM.Cerebral small vessel disease:insights and opportunities from mouse models of collagen IV-related small vessel disease and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy[J].Stroke,2014,45:1215-1221.
4 De Silva TM,Miller AA.Cerebral small vessel disease:targeting oxidative stress as a novel therapeutic strategy?[J].Front Pharmacol,2016,7:61.
5 Hachinski V.World Stroke Day 2008:"little strokes,big trouble"[J].Stroke,2008,39:2407-2420.
6 Thompson CS,Hakim AM.Living beyond our physiological means:small vessel disease of the brain is an expression of a systemic failure in arteriolar function:a unifying hypothesis[J].Stroke,2009,40:e322-330.
7 Craggs LJ,Yamamoto Y,Deramecourt V,et al.Microvascular pathology and morphometrics of sporadic and hereditary small vessel diseases of the brain[J].Brain Pathol,2014,24:495-509.
8 LADIS Study Group.2001-2011:adecade of the LADIS (Leukoaraiosis And DISability) Study:what have we learned about white matter changes and smallvessel disease?[J].Cerebrovasc Dis,2011,32:577-588.
9 Attwell D,Buchan AM,Charpak S,et al.Glial and neuronal control of brain blood flow[J].Nature,2010,468:232-243.
10 De Silva TM,Faraci FM.Microvascular dysfunction and cognitive impairment[J].Cell Mol Neurobiol,2016,36:241-258.
11 Fisher CM.Lacunes:small,deep cerebral infarcts[J].Neurology,1965,15:774-784.
12 Fisher CM.Lacunar strokes and infarcts:a review[J].Neurology,1982,32:871-876.
13 Fisher CM.Lacunar infarcts-a review[J].Cerebrovasc Dis,1991,1:311-320.
14 Mead GE,Lewis SC,Wardlaw JM,et al.Severe ipsilateral carotid stenosis and middle cerebral artery disease in lacunar ischaemic stroke:innocent bystanders?[J].J Neurol,2002,249:266-271.
15 Jackson CA,Hutchison A,Dennis MS,et al.Differing risk factor profiles of ischemic stroke subtypes:evidence for a distinct lacunar arteriopathy?[J].Stroke,2010,41:624-629.
16 Potter GM,Doubal FN,Jackson CA,et al.Lack of association of white matter lesions with ipsilateral carotid artery stenosis[J].Cerebrovasc Dis,2012,33:378-384.
17 Macdonald RL,Kowalczuk A,Johns L.Emboli enter penetrating arteries of monkey brain in relation to their size[J].Stroke,1995,26:1247-1250;discussion 1250-1251.
18 Bailey EL,McCulloch J,Sudlow C,et al.Potential animal models of lacunar stroke:a systematic review[J].Stroke,2009,40:e451-458.
19 Pantoni L,Garcia JH,Gutierrez JA.Cerebral white matter is highly vulnerable to ischemia[J].Stroke,1996,27:1641-1646;discussion 1647.
20 Petito CK,Olarte JP,Roberts B,et al.Selective glial vulnerability following transient global ischemia in rat brain[J].J Neuropathol Exp Neurol,1998,57:231-238.
21 Brun A,Englund E.A white matter disorder in dementia of the Alzheimer type:a pathoanatomical study[J].Ann Neurol,1986,19:253-262.
22 Millikan C,Futrell N.The fallacy of the lacune hypothesis[J].Stroke,1990,21:1251-1257.
23 Boiten J,Lodder J.Lacunar infarcts.Pathogenesis and validity of the clinical syndromes[J].Stroke,1991,22:1374-1378.
24 Lee HY,Oh BH.Aging and arterial stiffness[J].Circ J,2010,74:2257-2262.
25 Poels MM,Zaccai K,Verwoert GC,et al.Arterial stiffness and cerebral small vessel disease:the Rotterdam Scan Study[J].Stroke,2012,43:2637-2642.
26 Caplan LR,Hennerici M.Impaired clearance of emboli(washout) is an important link between hypoperfusion,embolism,and ischemic stroke[J].Arch Neurol,1998,55:1475-1482.
27 Diehl RR.Cerebral autoregulation studies in clinical practice[J].Eur J Ultrasound,2002,16:31-36.
28 Silberberg DH,Manning MC,Schreiber AD.Tissue culture demyelination by normal human serum[J].Ann Neurol,1984,15:575-580.
29 Lammie GA,Brannan F,Wardlaw JM.Incomplete lacunar infarction (Type Ib lacunes)[J].Acta Neuropathol,1998,96:163-171.
30 Schley D,Carare-Nnadi R,Please CP,et al.Mechanisms to explain the reverse perivascular transport of solutes out of the brain[J].J Theor Biol,2006,238:962-974.
31 Weller RO,Djuanda E,Yow HY,et al.Lymphatic drainage of the brain and the pathophysiology of neurological disease[J].Acta Neuropathol,2009,117:1-14.
32 Wardlaw JM,Sandercock PA,Dennis MS,et al.Is breakdown of the blood-brain barrier responsible for lacunar stroke,leukoaraiosis,and dementia?[J].Stroke,2003,34:806-812.
33 Hainsworth AH,Oommen AT,Bridges LR.Endothelial cells and human cerebral small vessel disease[J].Brain Pathol,2015,25:44-50.
34 Wardlaw JM,Doubal F,Armitage P,et al.Lacunar stroke is associated with diffuse blood-brain barrier dysfunction[J].Ann Neurol,2009,65:194-202.
35 Topakian R,Barrick TR,Howe FA,et al.Blood-brain barrier permeability is increased in normal-appearing white matter in patients with lacunar stroke and leucoaraiosis[J].J Neurol Neurosurg Psychiatry,2010,81:192-197.
36 Tomimoto H,Akiguchi I,Suenaga T,et al.Alterations of the blood-brain barrier and glial cells in white-matter lesions in cerebrovascular and Alzheimer's disease patients[J].Stroke,1996,27:2069-2074.
37 Utter S,Tamboli IY,Walter J,et al.Cerebral small vessel disease-induced apolipoprotein E leakage is associated with Alzheimer disease and the accumulation of amyloid beta-protein in perivascular astrocytes[J].J Neuropathol Exp Neurol,2008,67:842-856.
38 Young VG,Halliday GM,Kril JJ.Neuropathologic correlates of white matter hyperintensities[J].Neurology,2008,71:804-811.
39 Viggars AP,Wharton SB,Simpson JE,et al.Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology:a study in the MRC-CFAS population neuropathology cohort[J].Neurosci Lett,2011,505:25-30.
40 Heye AK,Thrippleton MJ,Chappell FM,et al.Blood pressure and sodium:Association with MRI markers in cerebral small vessel disease[J].J Cereb Blood Flow Metab,2016,36:264-274.
41 FredrikssonK,Nordborg C,Kalimo H,et al.Cerebral microangiopathy in stroke-prone spontaneously hypertensive rats.An immunohistochemical and ultrastructural study[J].Acta Neuropathol,1988,75:241-252.
42 Lin JX,Tomimoto H,Akiguchi I,et al.White matter lesions and alteration of vascular cell composition in the brain of spontaneously hypertensive rats[J].Neuroreport,2001,12:1835-1839.
43 Bailey EL,Wardlaw JM,Graham D,et al.Cerebral small vessel endothelial structural changes predate hypertension in stroke-prone spontaneously hypertensive rats:a blinded,controlled immunohistochemical study of 5- to 21-week-old rats[J].Neuropathol Appl Neurobiol,2011,37:711-726.
44 Bailey EL,McBride MW,Beattie W,et al.Differential gene expression in multiple neurological,inflammatory and connective tissue pathways in a spontaneous model of human small vessel stroke[J].Neuropathol Appl Neurobiol,2014,40:855-872.
45 Bragulat E,de la Sierra A,Antonio MT,et al.Endothelial dysfunction in salt-sensitive essential hypertension[J].Hypertension,2001,37:444-448.
46 Ma XJ,Cheng JW,Zhang J,et al.E-selectin deficiency attenuates brain ischemia in mice[J].CNS Neurosci Ther,2012,18:903-908.
47 Rouhl RP,Damoiseaux JG,Lodder J,et al.Vascular inflammation in cerebral small vessel disease[J].Neurobiol Aging,2012,33:1800-1806.
48 Aribisala BS,Wiseman S,Morris Z,et al.Circulating inflammatory markers are associated with magnetic resonance imaging-visible perivascular spaces but not directly with white matter hyperintensities[J].Stroke,2014,45:605-607.
49 Abbott NJ.In flammatory mediators and modulation of blood-brain barrier permeability[J].Cell Mol Neurobiol,2000,20:131-147.
50 Leong XF,Ng CY,Badiah B,et al.Association between hypertension and periodontitis:possible mechanisms[J].Scientific World Journal,2014,2014:768237.
51 Nakano K,Hokamura K,Taniguchi N,et al.The collagen-binding protein of Streptococcus mutans is involved in haemorrhagic stroke[J].Nat Commun,2011,2:485.
52 Miyatani F,Kuriyama N,Watanabe I,et al.Relationship between Cnm-positive Streptococcus mutans and cerebral microbleeds in humans[J].Oral Dis,2015,21:886-893.
53 Pussinen PJ,Alfthan G,Jousilahti P,et al.Systemic exposure to Porphyromonas gingivalis predicts incident stroke[J].Atherosclerosis,2007,193:222-228.
54 Lourbakos A,Yuan YP,Jenkins AL,et al.Activation of protease-activated receptors by gingipains from Porphyromonas gingivalis leads to platelet aggregation:a new trait in microbial pathogenicity[J].Blood,2001,97:3790-3797.
55 Walter C,Zahlten J,Schmeck B,et al.Porphyromonas gingivalis strain-dependent activation of human endothelial cells[J].Infect Immun,2004,72:5910-5918.
56 Ihara M,Yamamoto Y.Emerging evidence for pathogenesis of sporadic cerebral small vessel disease[J].Stroke,2016,47:554-560.
57 Laurent S,Briet M,Boutouyrie P.Large and small artery cross-talk and recent morbidity-mortality trials in hypertension[J].Hypertension,2009,54:388-392.
58 Scuteri A,Nilsson PM,Tzourio C,et al.Microvascular brain damage with aging and hypertension:pathophysiological consideration and clinical implications[J].J Hypertens,2011,29:1469-1477.
59 Bink DI,Ritz K,Aronica E,et al.Mouse models to study the effect of cardiovascular risk factors on brain structure and cognition[J].J Cereb Blood Flow Metab,2013,33:1666-1684.
60 Holland PR,Searcy JL,Salvadores N,et al.Gliovascular disruption and cognitive deficits in a mouse model with features of small vessel disease[J].J Cereb Blood Flow Metab,2015,35:1005-1014.
61 Nakaji K,Ihara M,Takahashi C,et al.Matrix metalloproteinase-2 plays a critical role in the pathogenesis of white matter lesions after chronic cerebral hypoperfusion in rodents[J].Stroke,2006,37:2816-2823.
62 Hattori Y,Okamoto Y,Maki T,et al.Silent information regulator 2 homolog 1 counters cerebral hypoperfusion injury by deacetylating endothelial nitric oxide synthase[J].Stroke,2014,45:3403-3411.
63 Feng S,Cen J,Huang Y,et al.Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins[J].PLoS One,2011,6:e20599.
64 Bailey EL,Smith C,Sudlow CL,et al.Is the spontaneously hypertensive stroke prone rat a pertinent model of sub cortical ischemic stroke? A systematic review[J].Int J Stroke,2011,6:434-444.
65 Corbin ZA,Rost NS,Lorenzano S,et al.White matter hyperintensity volume correlates with matrix metalloproteinase-2 in acute ischemic stroke[J].J Stroke Cerebrovasc Dis,2014,23:1300-1306.
66 Ihara M,Tomimoto H.Lessons from a mouse model characterizing features of vascular cognitive impairment with white matter changes[J].J Aging Res,2011,2011:978 761.
67 Armulik A,Abramsson A,Betsholtz C.Endothelial/pericyte interactions[J].Circ Res,2005,97:512-523.
68 Craggs LJ,Yamamoto Y,Ihara M,et al.White matter pathology and disconnection in the frontal lobe in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy(CADASIL)[J].Neuropathol Appl Neurobiol,2014,40:591-602.
69 Crag g s LJ,Fenwick R,Oak ley A E,et a l.Immunolocalization of platelet-derived growth factor receptor-β (PDGFR-β) and pericytes in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)[J].Neuropathol Appl Neurobiol,2015,41:557-570.
70 Bell RD,Winkler EA,Sagare AP,et al.Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging[J].Neuron,2010,68:409-427.
71 Miyamoto N,Maki T,Shindo A,et al.Astrocytes promote oligodendrogenesis after white matter damage via brain-derived neurotrophic factor[J].J Neurosci,2015,35:14 002-14 008.
72 Abbott NJ,Rönnbäck L,Hansson E.Astrocyteendothelial interactions at the blood-brain barrier[J].Nat Rev Neurosci,2006,7:41-53.
73 Maki T,Maeda M,Uemura M,et al.Potential interactions between pericytes and oligodendrocyte precursor cells in perivascular regions of cerebral white matter[J].Neurosci Lett,2015,597:164-169.
74 Nakane H,Ibayashi S,Fujii K,et al.Cerebral blood flow and metabolism in patients with silent brain infarction:occult misery perfusion in the cerebral cortex[J].J Neurol Neurosurg Psychiatry,1998,65:317-321.
75 Markus HS,Lythgoe DJ,Ostegaard L,et al.Reduced cerebral blood flow in white matter in ischaemic leukoaraiosis demonstrated using quantitative exogenous contrast based perfusion MRI[J].J Neurol Neurosurg Psychiatry,2000,69:48-53.
76 Fu JH,Lu CZ,Hong Z,et al.Relationship between cerebral vasomotor reactivity and white matter lesions in elderly subjects without large artery occlusive disease[J].J Neuroimaging,2006,16:120-125.
77 Shibata M,Ohtani R,Ihara M,et al.White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion[J].Stroke,2004,35:2598-2603.
78 Back SA,Han BH,Luo NL,et al.Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia[J].J Neurosci,2002,22:455-463.
79 Smith EE,Greenberg SM.Beta-amyloid,blood vessels,and brain function[J].Stroke,2009,40:2601-2606.
80 Vinters HV.Cerebral amyloid angiopathy.A critical review[J].Stroke,1987,18:311-324.
81 Vonsattel JP,Myers RH,Hedley-Whyte ET,et al.Cerebral amyloid angiopathy without and with cerebral hemorrhages:a comparative histological study[J].Ann Neurol,1991,30:637-649.
82 Zekry D,Duyckaerts C,Belmin J,et al.Cerebral amyloid angiopathy in the elderly:vessel walls changes and relationship with dementia[J].Acta Neuropathol,2003,106:367-373.
83 Smith EE,Gurol ME,Eng JA,et al.White matter lesions,cognition,and recurrent hemorrhage in lobar intracerebral hemorrhage[J].Neurology,2004,63:1606-1612.
84 Gurol ME,Irizarry MC,Smith EE,et al.Plasma betaamyloid and white matter lesions in AD,MCI,and cerebral amyloid angiopathy[J].Neurology,2006,66:23-29.
85 Holland CM,Smith EE,Csapo I,et al.Spatial distribution of white-matter hyperintensities in Alzheimer disease,cerebral amyloid angiopathy,and healthy aging[J].Stroke,2008,39:1127-1133.
86 Viswanathan A,Patel P,Rahman R,et al.Tissue microstructural changes are independently associated with cognitive impairment in cerebral amyloid angiopathy[J].Stroke,2008,39:1988-1992.
87 Viswanathan A,Greenberg SM.Cerebral amyloid angiopathy in the elderly[J].Ann Neurol,2011,70:871-880.
88 Kövari E,Charidimou A,Herrmann FR,et al.No neuropathological evidence for a direct topographical relation between microbleeds and cerebral amyloid angiopathy[J].Acta Neuropathol Commun,2015,3:49.
89 Brown WR,Moody DM,Challa VR,et al.Venous collagenosis and arteriolar tortuosity in leukoaraiosis[J].J Neurol Sci,2002,203-204:159-163.
90 Zhou M,Mao L,Wang Y,et al.Morphologic changes of cerebral veins in hypertensive rats:venous collagenosis is associated with hypertension[J].J Stroke Cerebrovasc Dis,2015,24:530-536.
91 Moody DM,Brown WR,Challa VR,et al.Periventricular venous collagenosis:association with leukoaraiosis[J].Radiology,1995,194:469-476.
92 Yan S,Wan J,Zhang X,et al.Increased visibility of deep medullary veins in leukoaraiosis:a 3-T MRI study[J].Front Aging Neurosci,2014,6:144.
93 Pantoni L,Gorelick PB.Cerebral small vessel disease[M].Cambridge,United Kingdom:Cambridge University Press,2014.
94 van Golen LW,Kuijer JP,Huisman MC,et al.Quantification of cerebral blood flow in healthy volunteers and type 1 diabetic patients:comparison of MRI arterial spin labeling and [(15)O]H2O positron emission tomography (PET)[J].J Magn Reson Imaging,2014,40:1300-1309.
95 Feng CM,Narayana S,Lancaster JL,et al.CBF changes during brain activation:fMRI vs.PET[J].Neuroimage,2004,22:443-446.
96 Wallin A,Blennow K,Fredman P,et al.Blood brain barrier function in vascular dementia[J].Acta Neurol Scand,1990,81:318-322.
97 Pantoni L,Inzitari D,Pracucci G,et al.Cerebrospinal fluid proteins in patients with leucoaraiosis:possible abnormalities in blood-brain barrier function[J].J Neurol Sci,1993,115:125-131.
98 Wallin A,Sjögren M,Edman A,et al.Symptoms,vascular risk factors and blood-brain barrier function in relation to CT white-matter changes in dementia[J].Eur Neurol,2000,44:229-235.
99 Taheri S,Gasparovic C,Shah NJ,et al.Quantitative measurement of blood-brain barrier permeability in human using dynamic contrast-enhanced MRI with fast T1 mapping[J].Magn Reson Med,2011,65:1036-1042.
100 Hansson J,Vasan RS,Ärnlöv J,et al.Biomarkers of extracellular matrix metabolism (MMP-9 and TIMP-1) and risk of stroke,myocardial infarction,and causespecific mortality:cohort study[J].PLoS One,2011,6:e16185.
101 Giwa MO,Williams J,Elderfield K,et al.Neuropathologic evidence of endothelial changes in cerebral small vessel disease[J].Neurology,2012,78:167-174.
102 Hu J,wanden Steen PE,Sang QX,et al.Matrix metalloproteinase inhibitors as therapy for in flammatory and vascular diseases[J].Nat Rev Drug Discov,2007,6:480-498.
