毛细管电泳法考察全氟辛酸与人血清白蛋白的相互作用
2018-01-11殷欣欣
顾 奕, 郭 明,*, 吕 达, 侯 平, 殷欣欣
(1. 浙江农林大学林业与生物技术学院, 浙江 杭州 311300; 2. 浙江农林大学理学院, 浙江 杭州 311300)
全氟化合物(perfluorinated compounds, PFCs)是一类新型有机污染物,主要包括全氟羧酸类(perfluorocarboxylic acids, PFCAs)、全氟磺酸类(perfluorinated sulfonic acids, PFSAs)和调聚醇类(fluorotelomer alchols, FTOHs)化合物等[1]。全氟化合物具有优良的热稳定性、化学稳定性、高表面活性及特有的疏水疏油性[2],被广泛用于食品包装材料中[3,4]。该类物质的C-F共价键具有极高的化学键能,使其普遍难以光解、水解、生物降解,因此多数全氟化合物具有环境持久性[5,6]。PFCs还具有生物累积性,可随着食物链的富集而聚集于人体中[7,8],且半衰期长,除了对人体肝脏、心脏、肺[9]等多种器官具有危害外,还具有神经[10]、免疫[11]、内分泌[12]毒性,严重威胁人类健康[13]。为此,美国食品药品监督管理局(FDA)于2016年禁止在食品接触材料中使用长链全氟化合物(单全氟烷基或双全氟烷基磷酸酯二乙醇胺盐、戊酸- 4,4- 双[(γ-ω- 全氟C8-20烷基)硫基]衍生物- 二乙醇胺化合物、磷酯酸的全氟烷基替代物)。全氟辛酸[14](pentadecafluorooctanoic acid, PFOA, C8HF15O2)是全氟化合物的典型代表,几乎涵盖了上述全部特点,是生态毒理学研究关注的热点之一。PFOA通过消化道的吸收、代谢作用进入血液循环后,经体液输运到达人体受体部位而发挥毒理作用,这与血浆的贮存和转运功能密不可分,而血清白蛋白是血浆中最丰富的载体蛋白,血浆对外来物质的载运功能主要由其承担[15]。因此,研究PFOA与血清白蛋白的相互作用可以在分子水平上认识PFOA与蛋白质作用的结合与载运机制和规律,有助于全面了解人血清白蛋白(HSA)- PFOA的互作机制和PFOA的毒理作用。然而,从文献报道来看,目前对PFOA在环境中的分布、迁移转化、生物富集、宏观整体水平毒理上[16,17]的研究较为集中,虽有利用荧光光谱技术研究PFOA与牛血清白蛋白相互作用的相关报道[18],但从分子水平角度探究PFOA的环境毒理作用还需进行深入研究,完善分析方法。因此研究PFOA与蛋白质相互作用的分析方法并构建PFOA与蛋白质相互作用的研究体系极有必要。
目前已有许多技术在分子水平研究污染毒物与生物大分子(如蛋白质、DNA、酶等)的相互作用,包括平衡透析法、超滤法、光谱技术、色谱法等[19]。相较而言,色谱法在研究相互作用的同时还能对复杂体系进行分离以减少共存化合物的干扰作用,而毛细管电泳除了具有色谱法的优点外,在模拟生理环境以维持生物介质活性方面更具优势,它具有应用范围广、分离效率高、分离模式多、检出限低等特点[20],已成功用于研究一些模型生物大分子与小分子之间的相互作用。如李玉娟等[21]利用毛细管电泳研究了丹参活性分子与牛凝血酶的相互作用;Zhou等[22]研究了苯二氮卓类药物氯氮平与HSA之间的相互作用。但从文献来看,毛细管电泳法在生物大分子与小分子相互的作用机制研究主要集中在酶、多肽等物质,与蛋白质的相互作用研究也主要集中于药物- 蛋白质体系,对蛋白质与重金属离子[23]、有机污染物[24]等小分子污染物的研究还处于上升阶段。
本文将构建PFOA- HSA相互作用体系,采用多种毛细管电泳应用模式:淌度移动法、区段- 区段动力学法(plug- plug kinetic, PPK法)、简化的Hummel- Dreyer (HD)法进行PFOA和HSA之间的研究,得到结合反应的相应参数,同时分析对比所得结果,从而建立分析PFOA- HSA相互作用的最适方法。相关结果可为进一步研究PFOA及其他全氟类化合物的毒性机理提供一定的理论参考。
1 实验部分
1.1 仪器、试剂与材料
P/ACETMMDQ毛细管电泳仪、未涂层熔融石英毛细管柱(60 cm×50 μm,有效长度50 cm)(美国贝克曼库尔特公司);旋转蒸发仪(上海申科科技有限公司); DF- 101S集热式恒温加热磁力搅拌器;KQ- 250DB数控超声波清洗器(昆山超声仪器有限公司); 1810D自动双重纯水蒸馏器(上海申生科技有限公司);分析天平(赛多利斯科学仪器(北京)有限公司);一次性使用无菌注射器(2.5 mL,圣光医用制品有限公司);微孔滤膜0.45 μm。
NaOH(批号20150310,含量≥96.0% )、Na2HPO4512H2O(批号20160219,含量≥99.0% )、NaH2PO452H2O(批号20151020,含量≥99.0% ),均购自国药集团化学试剂有限公司;PFOA(纯度90% , 英国Fluorochem公司);二甲苯类化合物(xylenes,批号EJ140079,纯度99% ,萨恩化学技术(上海)有限公司);苄胺(benzylamine, C7H9N,批号ED050140,纯度98% ,萨恩化学技术(上海)有限公司);氧化铝(Al2O3,中性,批号F20020726,中国医药集团(上海)化学试剂公司);盐酸(含量36% ~38% ,永华化学科技(江苏)有限公司);HSA(纯度96% ~99% ,美国Sigma公司);丙酮(分析纯,杭州双林化工试剂厂);N,N- 二甲基甲酰胺(DMF, HCON(CH3)2,批号20131122,含量≥99.5% ,国药集团化学试剂有限公司);实验用水为二次蒸馏水。
1.2 实验条件
1.2.1PFOA衍生反应
使用PPK法和简化的HD法进行测定时,需要以PFOA的光谱吸收值作为检测指标。由于PFOA没有特征紫外吸收谱,因此需要对其进行衍生反应,参考文献[25],对PFOA进行如下酰胺化衍生反应:

取PFOA 0.54 g、中性氧化铝0.08 g,加衍生化试剂(苄胺与二甲苯体积比为1∶2)12 mL,在140 ℃氮气保护条件下反应10 h,抽滤除去氧化铝。滤液通过旋转蒸发去除二甲苯和水后,加入10%盐酸20~30 mL至有固体析出,抽滤并用水冲洗,干燥得产物CF3(CF2)6COHNCH2ph。
1.2.2CE运行缓冲液及样品溶液的配制
配制0.2 mol/L Na2HPO4溶液(溶液A)和0.2 mol/L NaH2PO4溶液(溶液B),取81 mL溶液A与19 mL溶液B定容于1 000 mL容量瓶中,即配成pH 7.4的0.02 mol/L磷酸缓冲液。
称取0.103 5 g PFOA,用pH 7.4的磷酸缓冲液定容于25 mL容量瓶中,获得1×10-2mol/L PFOA母液。准确量取不同体积的PFOA母液,用pH 7.4磷酸缓冲液稀释至所需浓度((10、20、30、40、50、60、70、80、90、100)×10-5mol/L)(参考文献[26]中PFOA在人血清中的富集浓度进行配制),即配制成各浓度梯度的运行缓冲液。将上述溶液用0.45 μm微孔滤膜过滤两次并脱气5 min,备用。
以空白磷酸缓冲液配制1.0×10-5mol/L HSA溶液,加入5%(体积分数)丙酮[27](检测电渗流和迁移时间的重复性)混匀,用0.45 μm微孔滤膜过滤两次并脱气5 min,备用。
称取0.062 8 g PFOA衍生物,以pH 7.4磷酸缓冲液配制成5×10-3mol/L母液,精密移取不同体积母液,用pH 7.4磷酸缓冲液稀释至所需浓度((10、20、30、40、50、60、70、80、90、100)×10-5mol/L),即配制成各浓度梯度的PFOA衍生物样品溶液。将上述溶液用0.45 μm微孔滤膜过滤两次并脱气5 min,备用。
以空白磷酸缓冲液配制1.0×10-3mol/L、1.0×10-5mol/L的HSA样品溶液,以0.45 μm微孔滤膜过滤两次并脱气5 min,备用。
1.2.3PFOA- HSA体系相互作用方法的建立
实验前用0.1 mol/L NaOH、二次蒸馏水、空白缓冲液分别冲洗毛细管5 min,随后用相应的运行缓冲液分别冲洗5 min;实验中每两次运行测定程序间用0.1 mol/L NaOH、二次蒸馏水及运行缓冲液冲洗毛细管5 min,各梯度分别测定3次,求平均值。
淌度移动法 以含不同浓度的PFOA溶液作为运行缓冲液,以含有丙酮的HSA为测试样,进行淌度移动法测定。电泳条件:柱温25 ℃;压力进样3 447.4 Pa×3 s;分离电压15 kV;分离时间15 min;检测波长214 nm。
PPK法 方法分3组进样,前两组电压相同,第三组改变电压。电泳条件:运行缓冲液0.02 mol/L磷酸缓冲液(pH 7.4);柱温25 ℃;压力进样3 447.4 Pa×3 s;分离时间15 min;检测波长214 nm。分离电压:第一组、第二组进样时电压保持20 kV;第三组进样时,为保持蛋白质与污染物小分子结合过程的一致,先施加20 kV电压0.5 min,后调整电压为15 kV;为了消除电压变化对峰高的影响,实验中使用含适量体积分数的DMF溶液(以磷酸缓冲液配制)作为参比。进样次序依次为:DMF、HSA(1.0×10-5mol/L)、PFOA衍生物(20×10-5mol/L)(第一组进样时,HSA区段由磷酸缓冲液代替)。
简化的HD法 该方法在给定PFOA衍生物浓度条件下分两次进样,一次样品,一次空白,根据两次进样所得的实验数据来分析PFOA衍生物与HSA的相互作用。以含有不同浓度的PFOA衍生物为背景电解质,以空白磷酸缓冲液为空白测试样,以仅含HSA(1.0×10-3mol/L)的磷酸缓冲液为样品进样,进行简化的HD方法测试。电泳条件:柱温25 ℃;压力进样3 447.4Pa×15 s;分离电压15 kV;分离时间15 min;检测波长214 nm。

图 1 PFOA衍生物的紫外吸收图Fig. 1 UV spectrum of pentadecafluorooctanoic acid (PFOA) derivative
2 结果与讨论
2.1 PFOA衍生物的紫外图谱
由图1可以看出,PFOA的酰胺化产物CF3(CF2)6COHNCH2ph在214 nm处有适当吸收,且吸光度值适当,因此在使用PPK法和简化的HD法进行相互作用分析测定时,可选择214 nm作为检测波长。
2.2 PFOA- HSA相互作用体系的CE应用模式
CE研究HSA与PFOA分子相互作用时,PFOA与HSA的结合为非共价结合,且是可逆平衡过程,即:P+nMMnP,其中P代表HSA, M代表PFOA, MnP代表PFOA- HSA复合物。CE分离过程中涉及HSA和PFOA的结合、解离平衡以及组分的分离,由于游离的PFOA与HSA、HSA- PFOA复合物的质荷比不同,因而电泳迁移速率不同,由此可应用淌度移动法、PPK法、HD法分离可逆平衡体系中的游离PFOA、游离HSA和PFOA- HSA复合物,进而通过检测信号分析。
淌度移动法 毛细管中载体电解质为含有一定浓度PFOA的磷酸缓冲液,而进样样品为含有丙酮的HSA缓冲液,载体缓冲液中PFOA与样品中的HSA不断发生可逆平衡反应,PFOA与HSA结合形成PFOA- HSA复合物,而与丙酮基本没有相互作用。在电场驱动下,如果HSA、PFOA- HSA复合物迁移速率相近则将形成丙酮、HSA和复合物两个正峰(见图2)。

图 2 淌度移动法的毛细管电泳图Fig. 2 Capillary electropherogram of mobility method

图 3 区段- 区段动力学方法的毛细管电泳图Fig. 3 Capillary electropherograms of plug- plug kinetic method a. DMF- PBS- PFOA derivative (the first injection, 20 kV); b. DMF- HSA- PFOA derivative ( the second injection, 20 kV); c. DMF- HSA- PFOA derivative (the third injection, 15 kV). DMF: dimethylformamide; PBS: phosphate buffer; HSA: human serum albumin.
PPK法 PFOA衍生物与HSA在毛细管中发生相互作用。第一组进样会形成PFOA衍生物、DMF两个正峰,第二、三组进样均会形成PFOA衍生物、DMF、HSA和复合物3个正峰,但由于二者的分离电压不同,所以迁移时间会发生改变(见图3),具体理论可参考文献[28]。
简化的HD法 PFOA衍生物与HSA在毛细管内发生相互作用达可逆动态平衡,在分离系统中存在HSA、PFOA衍生物、复合物3条区带,在电泳图上出现一正一负两个峰(见图4)。其中负峰相应于溶液区段中PFOA衍生物浓度低于背景缓冲液中的部分,差值部分是与HSA结合了的PFOA衍生物的量。正峰相应于因具有相似迁移速率而未能分开的HSA与复合物,具体理论可参考文献[29]。

图 4 简化的HD法的毛细管电泳图Fig. 4 Capillary electropherogram of simplified Hummel- Dreyer method
2.3 PFOA- HSA相互作用的实验结果
利用淌度移动法、PPK法、简化的HD法研究PFOA- HSA体系的相互作用,并分析所得结果。
2.3.1淌度移动法
针对污染物PFOA小分子(配体M)- 蛋白质大分子(受体P)间的结合反应体系,可得到描述各物质间电泳淌度关系的相互作用反应模型,可表示如下:
nM+P→MnP
(1)
对于结合物MnP,其表观结合常数表达式为
(2)
受体P的平均电泳淌度(μ)变化值介于游离受体P(没有配体M存在)的电泳淌度(μP)与结合物MnP的电泳淌度(μMnP)之间,可表示为
(3)
经一系列推导转化,当n=1时,可得出公式(4),具体推导过程可参考文献[30]。
(4)
方程式(4)是以电泳淌度来表示的Scatchard线性方程式,以1/(μ-μp)对1/[M]进行线性拟合,所得直线的截距和斜率的比值即为表观结合常数KB。但是在实际拟合过程中,μ与M不存在线性关系,Bowser和Chen[31]强调将测得的数据进行非线性拟合,尤其是在合理的[M]范围内(通常配体M的浓度高于受体P浓度的10~100倍[32]),能够有效降低计算所得的结合常数误差。
由式(4)可知,不同的[M]对应不同的μ值,根据电泳图谱获得迁移时间,计算有效淌度的公式如下:
(5)
其中,μeff表示受体P的有效淌度,μapp表示其表观淌度,μeof表示电渗流淌度,Lt表示毛细管总长,Ld表示从进样端到检测窗口的有效长度,V表示毛细管两端的外加电压,t表示直接从电泳图谱中得到的受体P的迁移时间,teof表示丙酮的迁移时间。计算得到的μeof可作为式(4)中的μ(游离受体P的有效淌度或受体P的平均有效淌度)。将其对[M]进行非线性拟合,更符合实际情况,所得结果更精确。因而本研究工作中的KB通过非线性拟合获得。
按实验方法,测得不同浓度PFOA的电泳图谱,结果见图5。

图 5 PFOA和HSA相互作用的电泳图Fig. 5 Electropherograms of HSA in interaction with PFOA PFOA concentrations (mol/L): a. 10×10-5; b. 50×10-5; c. 80×10-5.
由图5可见,PFOA浓度的增加不能显著影响丙酮的迁移行为,即丙酮与PFOA之间没有明显的相互作用。随着运行缓冲液中PFOA浓度的增加,HSA峰的迁移时间变化也不明显,需进一步计算确定HSA与PFOA之间是否发生了相互作用。
实验中通过改变运行缓冲液中PFOA的浓度,可获得不同浓度下HSA及丙酮的迁移时间,根据式(5)可计算在不同浓度条件下的HSA平均有效淌度,测定结果见表1。

表 1 HSA在含不同PFOA浓度运行缓冲液中的有效淌度
μeff: effective mobility of HSA.
由表1可知,HSA的有效淌度与电渗流方向相反,且随着运行缓冲液中PFOA浓度的增加而增大,其值从-1.842 2×10-4cm2/(V·s)变化至-2.173 8×10-4cm2/(V·s),表明随着PFOA浓度的增加,PFOA- HSA之间有相互作用且相互作用逐渐增强。
根据表1数据,以PFOA的浓度为横坐标,相应的μeff为纵坐标,按式(4)进行非线性拟合,得到PFOA- HSA之间的非线性拟合曲线图(见图6)。

表 2 区段-区段毛细管电泳动力学参数Koff的考察(n=3)

表 3 区段-区段毛细管电泳动力学参数Kon的考察(n=3)
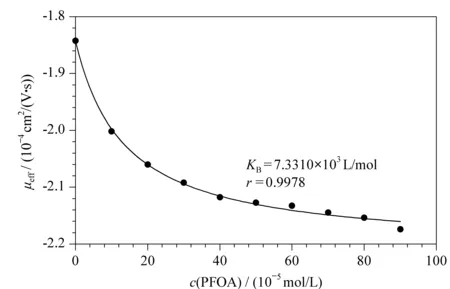
图 6 PFOA- HSA的非线性拟合曲线Fig. 6 Non- linear fitting curve of the PFOA- HSA
由图6可看出,除部分实验数据点偏离较大外,大多数能较好地靠近拟合曲线。经拟合,得到PFOA- HSA的表观结合常数KB为7.331 0×103L/mol。
2.3.2PPK法
污染物PFOA小分子与蛋白质的结合常数KB主要由结合速率常数Kon和解离速率常数Koff的比值决定。设物质A为PFOA衍生物,物质B为HSA,物质C为PFOA衍生物与HSA相互作用后的复合物PFOA衍生物- HSA。PFOA衍生物与HSA的动力学参数Kon、Koff和KB可由方程(6~8)得到:
(6)

(7)
(8)
本实验中,a0、b0、c1为发生相互作用后物质A、B、C的浓度;ha0对应于PFOA衍生物浓度为a0时峰高与DMF峰高的比值,ha2、ha3、hb0、hb2和hb3代表的含义以此类推,t2和t3分别为不同运行电压下PFOA衍生物或HSA的出峰时间,可在保证A与B结合过程一致的前提下通过改变电压获得,t1为A与B发生结合的时间。具体参数的计算公式可参考文献[33,34]。
对PFOA衍生物与HSA相互作用的电泳图谱(图3)的峰高进行计算,第一次进样由hPFOA衍生物/hDMF可获得参数ha0,第二次进样由hPFOA衍生物/hDMF可获得参数ha2,第三次进样由hPFOA衍生物/hDMF可获得参数ha3,进而根据式(6)、(7)即可求得Koff和Kon,详细参数见表2和表3。由式(8)求出PFOA衍生物和HSA相互作用结合常数KB:
KB=Kon/Koff=5.8835×103L/mol。
2.3.3简化的HD法
分别用背景缓冲液配制一系列不同浓度(10×10-5~100×10-5mol/L)的PFOA衍生物标准溶液,在相同条件下,得到不同浓度的PFOA衍生物的电泳图谱。以峰面积(A)对浓度(c, 10-5mol/L)进行线性回归,得到良好的线性关系。线性回归方程为A=1 137.8+360.15c,N=6, 相关系数(r)为0.998 1。
假设蛋白质与污染物分子的结合是理想状态,相互之间反应互不干扰、相互独立,那么其平衡关系可以用如下多级平衡方程式表示:
(9)
其中R为结合比,m为结合位点总类数,ni为第i类位点数,Ki为第i类位点的结合常数,Cb是结合小分子的浓度,Cf是游离小分子浓度,Cp是总蛋白浓度。Cf与R呈非线性关系,若小分子与所有结合位点的亲和力相同(m=1),方程(9)可变形为Cf与R的非线性方程
(10)
可将方程(10)简化为R与R/Cf的线性方程:
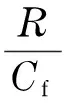
=-KBR+nKB (11)
For non-linear regression,y:R,x:Cf; For Scatchard,y:R/Cf,x:R; For Klotz,y: 1/R,x: 1/Cf.
方程(11)就是著名的Scatchard方程。在这个方程式中,根据斜率和截距即可直接求出结合常数KB和结合位点数n。Klotz提出了相互作用的Klotz线性方程式:
(12)
图7中PFOA衍生物的紫外吸收峰随着PFOA衍生物浓度的增加而逐渐增大,HSA和复合物的紫外吸收峰随着PFOA衍生物浓度的增加而呈下降趋势,说明PFOA衍生物和HSA发生了结合,形成了复合物。图7中PFOA衍生物的峰在HSA峰的前面,这是由于在PFOA衍生物和HSA的混合样中,试样中PFOA衍生物与HSA发生了可逆结合反应,形成了复合物。在本实验条件下,PFOA衍生物与HSA质荷比不同、HSA和复合物分子带负电荷,它们的电泳方向与电渗方向相反,由于电渗速率大于电泳速率,且PFOA衍生物的质荷比小于HSA与PFOA复合物的质荷比,故PFOA衍生物的迁移速率大于HSA与PFOA复合物,出峰于HSA之前,由于HSA与PFOA衍生物- HSA复合物的质荷比相近,HSA与复合物没有完全分开,所以会出现一个正峰。

图 7 简化的HD法测得的PFOA衍生物与HSA 相互作用电泳图Fig. 7 Electropherograms of HSA in interaction with PFOA derivative using simplified Hummel- Dreyer method
如表4所示,使用3种不同的方程(非线性回归方程、Scatchard方程、Klotz方程)构建PFOA衍生物- HSA相互作用模型,均能有效反映体系的作用效果: Klotz方程得到的结合常数KB值最大,Scatchard方程次之,非线性回归方程最小,但都相差不大,说明PFOA衍生物与HSA的结合强度适中;非线性回归方程得到的结合位点数n最大,Scatchard方程次之,Klotz方程最小,皆为1.0左右,说明PFOA衍生物与HSA作用只有单一类型的结合位点。综合比较,通过模型适用度分析得出非线性回归方程是PFOA衍生物- HSA体系的最适方程。
比较了不同方法测得的KB值(淌度移动法7.331 0×103L/mol, PPK法5.883 5×103L/mol,简化的HD法7.852 1×103L/mol),发现PFOA和PFOA衍生物与HSA相互作用的结合常数都是103数量级,且3个结果差距不大。因此我们推测衍生反应对相互作用影响较小,PFOA衍生物与HSA的相互作用可以近似认为是PFOA与HSA的相互作用。
2.3.4方法比较
用淌度移动法、PPK法和简化的HD法研究了全氟化合物PFOA与HSA的相互作用。经过对比发现3种方法均可获得结合常数KB,简化的HD法还可得到结合位点数n。PPK法实验操作简单,耗时短,适用于本体系的快速检测,作为辅助手段验证其他实验结果的有效性。简化的HD法出峰清晰,且随实验组PFOA衍生物浓度的递增,峰高出现明显的变化趋势:负峰(代表与HSA结合的PFOA衍生物)逐渐增高,正峰(代表游离蛋白质及复合物)逐渐降低。综合比较,淌度移动法耗时长,PPK法简单快捷,简化的HD法精准可靠。
2.3.5KB参数的生物学意义
利用简化的HD法获得的结合常数KB,根据Van’t Hoff方程,还可计算结合自由能变。
ΔG=-RTlnKB
(13)
其中R为气体常数(8.314 J/(mol5K)),KB为温度T(298 K)下的结合常数,由此算出结合自由能变ΔG为-22.22 kJ/mol,说明相互作用是自发进行的。
根据小分子与HSA结合生成的复合物的稳定性,结合反应过程的动力学特征,可将相互作用平衡体系粗略分为两种模型,一种具有慢动力学特征(KB数量级为103L/mol[35]),即蛋白质- 小分子复合物比较稳定。另一种能快速达到平衡(KB数量级为104~106L/mol[35]),其蛋白质- 小分子复合物的半衰期小于1 s。由实验结果可知,KB数量级为103L/mol,说明PFOA与HSA间的相互作用是慢平衡反应,表明PFOA与HSA的结合比较稳定,在体内消除慢,验证了PFOA的生物累积性。
3 结论
本工作应用毛细管电泳技术,将淌度移动法、PPK法、简化的HD法的理论用于实践研究,于分子水平上探究了环境污染物PFOA与HSA的相互作用,构建合理的理论模型并计算相关结合参数。简化的HD法运用了多个方程进行模型适用度分析,具有耗样量小、获得的结合信息较全面、分析快捷准确等优点,可反映PFOA衍生物与HSA的结合状况。本文的有关结果不仅对PFOA与HSA相互作用机制的进一步探究有重要意义,而且可为研究环境中其他类似的污染物- HSA体系的分子作用机理研究提供一定参考。需要说明的是,在配制样品浓度时是参考文献报道的PFOA在人血清中的富集浓度[26]进行的,达μg/L,与水中仅有的ng/L级别的浓度有较大差异。在极低浓度下PFOA与HSA的相互作用是否会有变化,还需后续实验进一步探讨。
[1] Wei L E, Shao M H, Zhang J, et al. Acta Scientiae Circumstantiae, 2016, 35(5): 1723
魏立娥, 邵秘华, 张晶, 等. 环境科学学报, 2016, 35(5): 1723
[2] Arvaniti O S, Andersen H R, Thomaidis N S, et al. Chemosphere, 2014, 111(111): 405
[3] Li F S, Ni H, Huang H Y, et al. Environmental Science, 2017, 38(1): 327
李法松, 倪卉, 黄涵宇, 等. 环境科学, 2017, 38(1): 327
[4] Dong X Y, Fan L J, Zhang Z L, et al. Toxicology, 2017, 380: 23
[5] Santos A, Rodríguez S, Pardo F, et al. Sci Total Environ, 2016, 563/564: 657
[6] Rainieri S, Conlledo N, Langerholc T, et al. Food Chem Toxicol, 2017, 104: 14
[7] Müller C E, De Silva A O, Small J, et al. Environ Sci Technol, 2011, 45(20): 8665
[8] Xu J, Guo C S, Zhang Y, et al. Environ Pollut, 2014, 184(1): 254
[9] Cui L, Zhou Q F, Liao C Y, et al. Arch Environ Contam Toxicol, 2009, 56(2): 338
[10] Mariussen E. Arch Toxicol, 2012, 86(9): 1349
[11] Corsini E, Luebke R W, Germolec D R, et al. Toxicol Lett, 2014, 230(2): 263
[12] Crawford N M, Fenton S E, Strynar M, et al. Reprod Toxicol, 2017, 69: 53
[13] Lau C. Perfluorinated Compounds: an Overview. Basel, Switzerland: Springer International Publishing, 2015
[14] Zhu L Y, Lin J H. Chinese Journal of Applied Ecology, 2008, 19(5): 1149
祝凌燕, 林加华. 应用生态学报, 2008, 19(5): 1149
[15] Pahari B P, Chaudhuri S, Chakraborty S, et al. J Phys Chem B, 2015, 119(6): 2533
[16] Arias E V A, Mallavarapu M, Naidu R. Environ Monit Assess, 2009, 43(24): 9723
[17] Mortensen A S, Letcher R J, Cangialosi M V, et al. Chemosphere, 2011, 83(8): 1035
[18] Xie X C, Wang X R, Zhang Y K, et al. China Environmental Science, 2010, 30(11): 1496
谢显传, 王晓蓉, 张幼宽, 等. 中国环境科学, 2010, 30(11): 1496
[19] Bi C, Jackson A, Vargas- Badilla J, et al. J Chromatogr B, 2016, 1021: 188
[20] Rundlett K L, Armstrong D W. Electrophoresis, 2001, 22(7): 1419
[21] Li Y J, Wu G X, Ou W L, et al. Chinese Journal of Chromatography, 2017, 35(3): 339
李玉娟, 武广霞, 欧婉露, 等. 色谱, 2017, 35(3): 339
[22] Zhou D W, Li F M. J Pharm Biomed Anal, 2004, 35(4): 879
[23] Fan Y P, Li S, Fan L Y, et al. Chinese Journal of Chromatography, 2012, 30(8): 827
范银苹, 李杉, 樊柳荫, 等. 色谱, 2012, 30(8): 827
[24] Li T, Wang H. Anal Chem, 2009, 81(5): 1988
[25] Chen X D, Feng X, Zhang X D, et al. Applied Chemical Industry, 2006, 35(1): 33
陈晓东, 冯霄, 张向东, 等. 应用化工, 2006, 35(1): 33
[26] Scott B, Antonia C, Christopher L, et al. Epidemiol, 2009, 20(44): S255
[27] Zhou D W, Wang H F, L M F. Journal of China Pharmaceutical University, 2005, 36(1): 36
周大炜, 王怀锋, 李发美. 中国药科大学学报, 2005, 36(1): 36
[28] Okhonin V, Petrov A P, Berezovski M, et al. Anal Chem, 2006, 78(14): 4803
[29] Guo M, Fan W X, Feng C M, et al. Chinese Journal of Biochemistry and Molecular Biology, 2014, 30(10): 1039
郭明, 范文翔, 冯春苗, 等. 中国生物化学与分子生物学报, 2014, 30(10): 1039
[30] Guo M, Liu M M, Li M H, et al. Chinese Journal of Analytical Chemistry, 2012, 40(2): 268
郭明, 刘咪咪, 李铭慧, 等. 分析化学, 2012, 40(2): 268
[31] Bowser M T, Chen D. J Phys Chem A, 1998, 103(1): 197
[32] Zhang Y Z, Zhou B, Liu Y X, et al. J Fluoresc, 2008, 18(1): 109
[33] Liu C Y, Zhang X J, Miao Y Q, et al. Journal of Analytical Science, 2015, 31(3): 313
刘春叶, 张雪娇, 苗延青, 等. 分析科学学报, 2015, 31(3): 313
[34] Xiong C Q, Xia Z N, Huang R, et al. Scientia Sinica Chimica, 2008, 38(8): 705
熊彩侨, 夏之宁, 黄锐, 等. 中国科学(B辑: 化学), 2008, 38(8): 705
[35] Heegaard N H, Nissen M H, Chen D D. Electrophoresis, 2002, 23(6): 815
