Preparation of monolithic fibers with deep eutectic solvent for solid phase microextraction of polycyclic aromatic hydrocarbons in lake water
2018-01-11CHENNaZHANGYijunZHAOWanliCHENJunZHANGYuping
CHEN Na, ZHANG Yijun, ZHAO Wanli, CHEN Jun, ZHANG Yuping
(School of Chemistry and Chemical Engineering, Henan Institute of Science andTechnology, Xinxiang 453003, China)
Within the framework of green chemistry, solvents occupy a strategic position. To qualify as a green medium, solvents have to be readily available, non- toxic, biodegradable, recyclable, non- flammable, and inexpensive [1]. Deep eutectic solvents (DESs) are a rapidly emerging family of ionic fluids [2,3]. They were developed by Abbott et al. [4] in 2003, and are generally composed of two or three cheap and non- toxic components that can self- associate through hydrogen bond interactions to form a eutectic mixture with a melting point lower than that of its individual components [5,6]. DESs are generally liquid at temperatures lower than 100 ℃ and exhibit similar physicochemical properties to ionic liquids, while being much cheaper and more environmentally friendly [7]. Because of these advantages, interest in DESs is growing in many fields.
Gu et al. [8] used DESs as extractants to concentrate polar compounds (phenols, amines, and acids) from non- polar solvents using an ultrasonic wave- assisted liquid- phase microextraction (LPME) method. Li et al. [9]used DESs to prepare molecularly imprinted polymers for rapid purification of caffeic acid from hawthorn with solid phase extraction (SPE). In another study [10], ternary DESs were used as a functional monomer in molecularly imprinted polymers for purification of levofloxacin. Zhu et al. [11] extracted chitin with high purity from lobster shells using choline chloride- malonic acid DESs.
Solid phase microextraction (SPME) is a sample preparation technique that integrates sampling, isolation, enrichment, and injection in one step [12-14]. SPME has sparked great interest in the scientific community because it is solvent- free, sensitive, fast, simple, and easily incorporated into chromatographic analysis. The commercialized SPME fiber has a layer coated on the surface of a quartz fiber [15,16], and is fragile and short- lived. Thus, it would be of interest to prepare a SPME fiber using other materials. Our team have used monolithic materials in capillaries to prepare monolithic fibers [17-19]. But, the porogens were organic solvents, which were toxic and environmental pollution. Thus, it’s necessary to prepare a fiber using non- toxic, environmental friendly and cheap reagents.
The aim of the present study was to use a DES as a porogen for the preparation of monolithic SPME fibers. Instead of traditional organic solvents, DESs were used to prepare a poly(butyl methacrylate- ethylene glycol dimethacrylate) [poly(BMA- EDMA)] monolithic fiber by microwave irradiation. The operating conditions affecting extraction of the target polycyclic aromatic hydrocarbons (PAHs) by SPME combined with ultra performance liquid chromatography (UPLC) were optimized. The method was then applied to the determination of the three PAHs (naphthalene, biphenyl, and phenanthrene) in lake water.
1 Experimental
1.1 Apparatus and chemicals
UPLC analysis was performed with an Agilent 1290 Infinity II Series UPLC system (Agilent Technologies, USA). Fused silica capillaries (530 μm inner diameter) were purchased from Yongnian Photoconductive Fiber Factory (Hebei, China). Irradiation experiments were carried out using a domestic microwave oven (Midy Co. Ltd, Fo- shan, China) with a microwave power of 700 W and a frequency of 2 450 Hz. The SPME equipment were purchased from Supelco (USA), including 4- mL vials, 200- μL vials, and the commercial polydimethylsiloxane (PDMS) fiber (100 μm). A magnetic stirrer (TWCL- D, Henan Aibote Technology Development Co., China) was applied. The morphologies of the monolithic materials were characterized using a Quanta 200 scanning electron microscope (FEI, USA).
The DES porogen was prepared by mixing choline chloride (purity: 98% ) and ethanediol (purity: 98% , Aladdin Industrial Corporation, Shanghai, China) in molar ratio of 1∶2. Butyl methacrylate (BMA), ethylene glycol dimethacrylate (EDMA), and azobisisobutyronitrile (AIBN) were purchased from J & K Scientific Ltd. (Beijing, China). BMA and EDMA were extracted with 5%-10% (mass fraction) sodium hydroxide aqueous solution and water before use, respectively. High performance liquid chromatography (HPLC) grade acetonitrile (ACN) and methanol (MeOH) were purchased from Merck (Germany). Naphthalene, biphenyl, and phenanthrene were purchased from Aladdin Industrial Corporation (Shanghai, China). Sodium hydroxide (NaOH), sodium chloride, acetone,n- hexane,n- butanol, isopropanol (ISO), and ethyl acetate (EtOAc) were purchased from Tianjin Kemi’ou Chemical Reagent Company, China. All reagents were of analytical grade and were used without further purification.
1.2 Preparation of standard solutions
The three standard PAHs were dissolved in methanol to give a mass concentration of 100 mg/L. A series of standard solutions were prepared at different mass concentrations in the range of 0.1-8.0 mg/L, which was dissolved in water. The standard curves of the three PAHs were linear by assaying 14 data points and each sample was determined for three times.
1.3 Preparation of the poly(BMA- EDMA) monolithic fiber
A simple and convenient in- situ polymerization technique was used for preparation of the poly(BMA- EDMA) monolithic fiber. First, a 10- cm capillary was flushed with acetone, methanol, and ultrapure water each for 30 min, respectively, and then dried with air. A polymerization of 0.6 mL of BMA monomer, 0.9 mL of EDMA crosslinker, 1.5 mL ofn- butanol, and 0.5 mL of DES porogen was mixed to form a homogeneous solution, and then ultrasonicated for about 10 min. AIBN (0.02 g) was added as a polymerization initiator. The solution was degassed thoroughly by ultrasonication for about 10 min, purged with nitrogen gas for 30 min, and then introduced into the capillary. Each end of capillary was then sealed with a small piece of rubber. The filled capillary was placed in a domestic microwave oven for 4 min at 100% power. After the polymerization, 1 cm of the monolith in capillary was pushed out with a stainless steel wire (300 μm in diameter). Finally, the fibers were immersed in 5 mL of methanol with agitation (200 r/min) for about 1 h. This procedure was repeated six times after replacing methanol until no compounds (unreacted monomers and porogens) were detected by UPLC. The same method was used to prepare a fiber without the DES.
1.4 SPME procedure
The SPME procedure involved extraction and desorption. First, the sample solution (4 mL) was placed in a 4- mL vial containing a stirring bar (8 mm×2 mm). The extraction was performed by direct immersion of the fiber in the sample solution for 30 min with slow stirring (200 r/min). After the extraction, the fiber was removed and desorbed with acetonitrile in a 200- μL vial for 20 min. All experiments were performed at room temperature.
1.5 UPLC analysis
A UPLC analysis was performed on a Zorbax RRHD C18column (50 mm×2.1 mm, 1.8 μm, Agilent Technologies, USA). The mobile phases were acetonitrile- water (50∶50, v/v). The flow rate was 1.0 mL/min, and the detection wavelength was 210 nm. The injection volume was 1 μL.
2 Results and discussion
2.1 Comparison of the fibers with and without DES and the PDMS fiber

Fig. 1 Comparison of the extraction efficiencies among fibers with DES, without DES, and a commercial PDMS fiber (100 μm)
To investigate the performance of DES as a porogen, monolithic fibers were prepared with and without DES. Both these fibers were compared with a commercial PDMS (100 μm) fiber (Fig. 1). For the three PAHs, the extraction efficiencies obtained by poly(BMA- EDMA) fiber with the DES were two times than those of the fiber without DES. The extraction efficiency for naphthalene by the fiber with the DES was about four times than that of the PDMS fiber.
2.2 Morphologies of the fibers with and without DES
The fibers with and without DES by in- situ polymerization were prepared and their morphologies were observed by scanning electron microscopy (SEM) (Fig. 2). Both fibers were integrated and homogeneous. The fiber with DES (Fig. 2a) had a clearer skeleton and larger pore sizes than the fiber prepared without DES (Fig. 2b), and these pores could be advantageous for extraction and desorption.

Fig. 2 Scanning electron micrographs of the fibers prepared (a) without DES and (b) with DES
2.3 Optimization of the extraction conditions
To optimize the extraction performance, the effects of the extraction solvent, extraction time, desorption solvent, desorption time, and ionic strength of the sample matrices on the SPME were investigated in detail.
2.3.1Effects of the extraction solvent and extraction time
Because the extraction solvent could affect the efficiency of the SPME, we investigated several solvents (water, methanol, acetonitrile, ethyl acetate,n- hexane and isopropyl alcohol) under the same experimental conditions (Fig. 3a). Among these solvents, water provided the best extraction efficiency. Thus, the prepared fiber could be used to extract the PAHs in water matrix, like lake water.
The extraction time was optimized in the range of 1-90 min for the three PAHs (Fig. 3b). The extraction efficiency increased as the extraction time was increased up to 30 min, and then remained constant. Therefore, 30 min was sufficient time to allow for the extraction to reach equilibrium. This is presumable the balance point between the amount extracted and the extraction time.

Fig. 3 Effects of the (a) extraction solvents, (b) extraction times, (c) desorption solvents, and (d) desorption times on the peak area of the three PAHs
2.3.2Effects of the desorption solvent and desorption time
In SPME, complete elution of the target compounds depends on selection of a suitable desorption solvent. We evaluated ACN, MeOH, ACN- H2O (85∶15, v/v), MeOH- H2O (85∶15, v/v), and water as desorption solvents under the same experimental conditions (Fig. 3c). The highest extraction efficiencies were achieved with ACN and MeOH- H2O (85∶15, v/v). ACN was selected to be the desorption solvent since it was used as the mobile phase.
The effect of desorption time (1-50 min) was also investigated (Fig. 3d). Desorption of the three target PAHs increased when the desorption time was increased from 1 to 20 min, and then remained almost constant with further increases in the desorption time from 20 to 50 min. Therefore, 20 min was selected for desorption in subsequent experiments.
2.3.3Effect of ionic strength
Generally, as the ionic strength of a sample matrix increases, the extraction is affected by salting out and electrostatic interactions [20]. These act in opposition with salting out improving the extraction performance and electrostatic interactions between polar molecules and salt ions in the sample solution reducing the extraction efficiency.
The effect of the ionic strength of the matrix was investigated by addition of NaCl at various concentrations (0, 10, 50, and 100 g/L). The electrostatic interaction effect was the predominant of these two effects over the entire ionic strength range tested, which meant that the extraction efficiencies of the three PAHs decreased with increasing ionic strength. Therefore, addition of NaCl to the extraction solution was not necessary for subsequent experiments.
2.4 Linear ranges, LODs, and RSDs
The analytical performance of the method was evaluated under the optimized conditions (Table 1). The linear ranges of the three PAHs were 0.1-6.0 mg/L with correlation coefficients (r) ≥0.990 3. The limits of detection (LODs,S/N=3) were in the ranges of 2.1-4.9 μg/L. Using the same experimental conditions, a DES fiber was used six times in a row. The relative standard deviations (RSDs) of the retention times for the three PAHs were in the range of 11.2% -15.1% . six DES fibers, the RSDs of the retention times were between 10.6% and 13.1% . Preparation of a DES fiber for SPME showed good precision, and could meet the needs of daily analysis.

Table 1 Linear equations, r, linear ranges, LODs and RSDs of retention times for the three PAHs
Y: peak area;X: mass concentration, mg/L.
2.5 Analysis of real samples
This method was applied to the determination of the three target PAHs in lake water samples. None of the PAHs were detected in the lake water samples. For validation of the extraction efficiency, lake water samples spiked with 1 mg/L PAHs before and after extraction were analyzed and compared (Fig. 4). The PAHs were obviously enriched after spiking, with an enrichment factor of >10.7 (the ratio of peak area) for naphthalene. The accuracy of the method was evaluated using the recoveries of the PAHs from samples spiked with 0.2, 0.3, and 0.4 mg/L. The recoveries of the three PAHs from lake water samples were in the range of 86.4% -111.3% (Table 2). All RSDs (n=3) of the recoveries were ≤ 8.0% .

Fig. 4 Chromatograms of the three PAHs (1 mg/L) spiked in a blank lake water sample by direct sampling and SPME
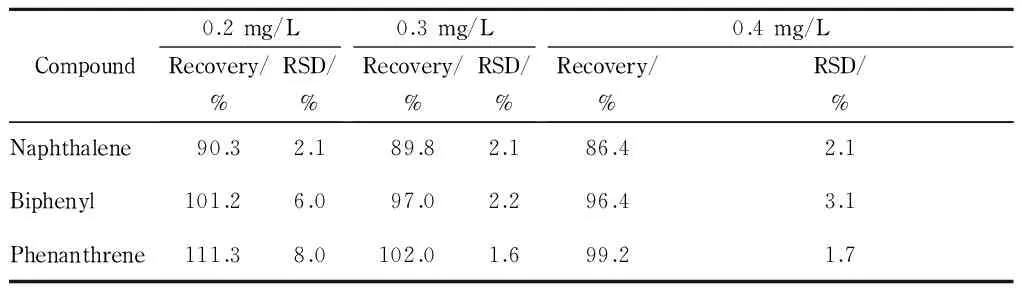
Compound0.2mg/LRecovery/%RSD/%0.3mg/LRecovery/%RSD/%0.4mg/LRecovery/%RSD/%Naphthalene90.32.189.82.186.42.1Biphenyl101.26.097.02.296.43.1Phenanthrene111.38.0102.01.699.21.7
3 Conclusions
In this work, a monolithic fiber with DES as a porogen was prepared for SPME and used for effective extraction of the three PAHs from lake water. Preparation of the DES fiber by in- situ polymerization is simple and reproducible, and the DES monolithic fiber can be used for effective extraction of the PAHs. The method is suitable for the analysis of the PAHs in lake water, and is inexpensive, environmentally friendly, highly sensitive, and reproducible.
[1] Gu Y L, Jerome F. Chem Soc Rev, 2013, 42(24): 9550
[2] de Faria E L P, do Carmo R S, Claudio A F M, et al. Int J Mol Sci, 2017, 18(11): 2276
[3] Hou X D, Li A L, Lin K P, et al. Bioresour Technol, 2018, 249: 261
[4] Abbott A P, Capper G, Davies D L, et al. Chem Commun, 2003, 1(1): 70
[5] Zhang Q H, Vigier K D O, Royer S, et al. Chem Soc Rev, 2012, 41(21): 7108
[6] Kudlak B, Owczarek K, Namiesnik J. Environ Sci Pollut Res, 2015, 22(16): 11975
[7] Ali I, Suhail M, Sanagi M M, et al. Crit Rev Anal Chem, 2017, 47(4): 332
[8] Gu T N, Zhang M L, Tan T, et al. Chem Commun, 2014, 50(79): 11749
[9] Li G Z, Tang W Y, Cao W M, et al. Chinese Journal of Chromatography, 2015, 33(8): 792
[10] Li X X, Row K H. J Chromatogr B, 2017, 1068/1069: 56
[11] Zhu P, Gu Z J, Hong S, et al. Carbohyd Polym, 2017, 177: 217
[12] Chen L M, Zhao M B, Liu H W. Journal of Instrumental Analysis, 2011, 30(8): 835
[13] Mei M, Huang X J. Chinese Journal of Chromatography, 2016, 34(12): 1168
[14] Wang N N, Shou D, Wang X P, et al. Chinese Journal of Chromatography, 2017, 35(1): 14
[15] Bai J Y, Huang R H, Lu Y M, et al. Chinese Journal of Chromatography, 2016, 34(8): 778
[16] Abdulra’uf L B, Hammed W A, Tan G H. Crit Rev Anal Chem, 2012, 42(2): 152
[17] Jin Y F, Chen N, Liu R Q, et al. J Chin Chem Soc, 2013, 60(8): 1043
[18] Chen J, Bai L Y, Tian M K, et al. Anal Methods, 2014, 6(2): 602
[19] Chen N, Zhang Y J, Chen J, et al. Chinese Journal of Analysis Laboratory, 2013, 32(8): 53
[20] Cui M Y, Qiu J X, Li Z H, et al. Talanta, 2015, 132: 564
