活性炭固定床吸附分离溶液中表面活性剂和多环芳烃
2018-01-08温舒晴赵思维刘剑飞
刘 凡,温舒晴,王 昌,赵 营,赵思维,刘剑飞
(河南理工大学 土木工程学院,河南 焦作 454003)
多环芳烃(PAH)是一类典型的长期性有机污染物,具“三致”作用,在土壤基质中经迁移转化成为地下水的长期污染源,故其污染场所的修复受到了环境学界的广泛关注[1-3]。PAH属疏水性有机物,需投加表面活性剂以降低土壤中PAH的界面张力,增加其水溶解性和生物有效性,从而提高污染土壤的修复效果[4-6]。但表面活性剂的消耗也增加了土壤修复的成本,其费用可达总运行费用的50%[7],故有必要对表面活性剂进行分离回收并加以再利用。目前的分离技术有气提法[8-10]、液液萃取法[11]、冷却沉淀法[12]、光化学分解法[13]、离子交换法[14]、臭氧氧化法[15]、膜处理法[16]、反胶团处理法[17]等,这些方法存在限制条件多、分离不完全、能耗大、成本高等问题[18],而吸附法是一种低耗能、低成本且应用广泛的技术。
目前使用活性炭吸附分离表面活性剂和疏水性有机物的研究非常有限,不多的研究也多是批处理实验[19-21],类似于工程中将活性炭投至废水池中,通过搅拌混合以达到良好的吸附效果,但需增加搅拌设备和分离设施。工业过程中多使用固定床吸附[22],可实现连续稳定运行,且无需专门的分离装置,但少有吸附分离两种不同物质的情况。Yang等[23]证实了使用活性炭固定床分离PAH和表面活性剂的可行性,但未见进一步的研究。
本工作选取具代表性的PAH和表面活性剂,利用活性炭固定床吸附分离表面活性剂和PAH的混合溶液,分析了活性炭填充量、PAH和表面活性剂浓度、流量和多溶质等因素对分离效果的影响,采用Thomas和BDST模型对吸附过程进行了模拟,并进行了成本分析,以期为工程设计提供理论基础。
1 实验部分
1.1 试剂和材料
Triton X-100(TX100):购于中国百灵威科技有限公司,非离子型表面活性剂,分析纯;菲(PHE)、荧蒽(FLA)、苯并蒽(BaA):购于日本东京化成工业株式会社,分析纯。
活性炭:购于中国西陇化工股份有限公司,比表面积718.2 m2/g,孔体积0.845 cm3/g,孔径分布0.2~2.0 nm,平均孔径0.845 nm,微孔体积0.397 cm3/g,中孔体积0.206 cm3/g,大孔体积0.242 cm3/g。
1.2 实验装置和方法
实验装置示意见图1。

图1 实验装置示意
固定床有机玻璃柱(自制)高20 cm,内径2.5 cm。固定床出水直接流入储槽中。在TX100和单种PAH体系中:配制6 L TX100和PHE的共存溶液于广口玻璃瓶中,控制流量、活性炭填充量、PHE质量浓度、TX100质量浓度等条件,按照设定的流量调节出水,待出水水流稳定后开始计时,每隔15 min取样。在TX100和多种PAH体系中:TX100质量浓度为5 g/L,PHE,FLA,BaA的质量浓度分别为120,80,12 mg/L。
1.3 分析方法
取水样静置30 min,用玻璃注射器取上清液,经0.2 μm聚四氟乙烯过滤器过滤至2 mL进样瓶中。采用Dionex公司U3000型高效液相色谱仪测定TX100和PAH的质量浓度,色谱柱为Agilent公司PAH柱(250 mm×4.6 mm),流动相(乙腈-水)体积比85∶15,柱温30 ℃,流量1 mL/min,检测UV波长230 nm。
2 结果与讨论
2.1 穿透曲线的影响因素
2.1.1 流量
在活性炭填充量20 g、初始PHE质量浓度120 mg/L、初始TX100质量浓度5 g/L的条件下,流量对穿透曲线的影响见图2,图中:t为吸附时间,min;ρ0和ρt为吸附质在初始和t时刻时的质量浓度,mg/L。
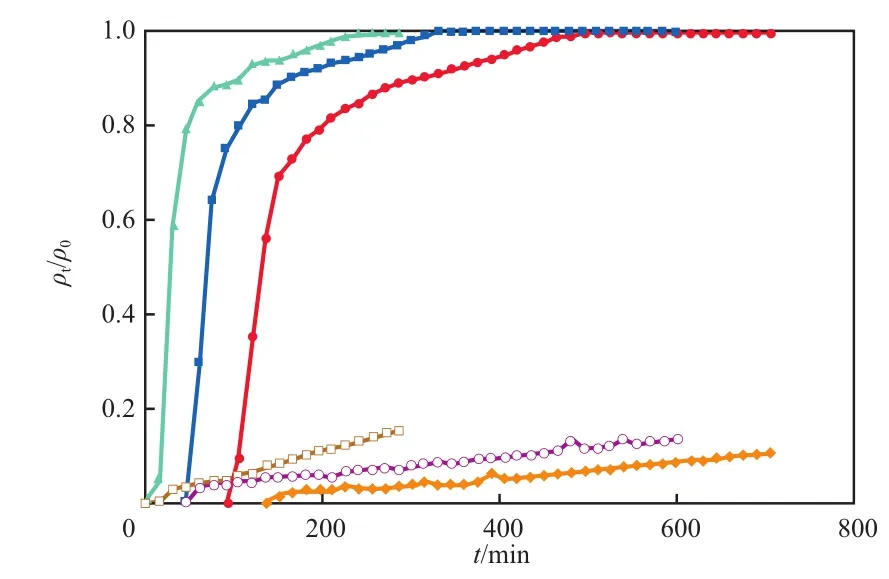
图2 流量对穿透曲线的影响
由图2可见:流量越高,TX100和PHE的穿透曲线的斜率越大,其穿透时间(ρt/ρ0=0.1时的时间)越短,因为流量越低,水力停留时间越长,溶液有更长的时间与活性炭接触[24];对应于流量30,20,10 mL/min,TX100的穿透时间分别为19,64,107 min,而吸附饱和时间(ρt/ρ0=0.9时的时间)分别为88,116,150 min,PHE的穿透时间分别为193,420,687 min。可见TX100的吸附饱和时间要短于PHE的穿透时间,这种现象正是我们所期待的,这为二者的分离提供了条件。
2.1.2 活性炭填充量
在流量20 mL/min、初始PHE质量浓度120 mg/L、初始TX100质量浓度5 g/L的条件下,研究活性炭填充量对穿透曲线的影响可知,随着填充量的增加,TX100和PHE的穿透时间均延长。此外,随着填充量的增加,活性炭柱的高度增高(填充量为10,15,20 g时对应的吸附柱高分别为7.0,10.8,14.7 cm),活性炭的吸附面积增大,使得TX100和PHE的吸附饱和时间缩短[25]。
2.1.3 PHE质量浓度
在流量20 mL/min、活性炭填充量20 g、初始TX100质量浓度5 g/L的条件下,研究初始PHE质量浓度对穿透曲线的影响可知,初始PHE的浓度越大其穿透时间越短,初始PHE质量浓度为60,90,120 mg/L时的穿透时间分别为584,502,420 min。这是由于浓度低则浓度梯度低,扩散作用小,故需要更长的穿透时间。另一方面,初始PHE的浓度变化对TX100的穿透时间影响并不大。这是由于二者的浓度差别较大,TX100的浓度大,扩散作用也大[26],故其穿透时间不易受到低浓度PHE的影响。
2.1.4 TX100质量浓度
在流量20 mL/min、活性炭填充量20 g、初始PHE质量浓度60 mg/L的条件下,研究初始TX100质量浓度对穿透曲线的影响可知,随着初始TX100浓度的增加,其穿透时间也延长,初始TX100质量浓度为3,4,5 g/L时对应的穿透时间分别为211,192,166 min。这主要也是由于浓度影响扩散作用,进而影响穿透时间所致。而初始TX100浓度的变化对PHE穿透时间的影响则不显著。
2.2 多PAH体系
由于实际工程中必然是多种物质共存,故需考虑多种PAH共存的情况。在流量20 mL/min、活性炭填充量20 g的条件下,多PAH体系的穿透曲线见图3。由图3可见:混合体系溶液流出活性炭固定床时,TX100首先穿透,然后是3种PAH穿透;对TX100而言,多种PAH的加入并未影响到其穿透时间,接近饱和的时间为150 min;对于3种PAH,其穿透曲线形状类似,只是穿透时间不同,PHE,FLA,BaA的穿透时间分别约为420,520,700 min。实验结果表明,由于表面活性剂和有机污染物的不同浓度和性质,导致其穿透时间不同,这样就创造了一段可以回用表面活性剂的时间。而在多PAH系统中,只要有一种污染物浓度超出其控制浓度时,就应该停止表面活性剂的回收。

图3 多PAH体系的穿透曲线
2.3 有效回收时间
从穿透曲线看,有一个共同规律,即浓度高的先穿透而浓度低的后穿透,这种现象为分离表面活性剂和PAH提供了条件。在TX100到达穿透时间前,由于溶液中的表面活性剂浓度较低,溶液不能用于土壤修复;当PHE到达穿透时间后,由于溶液中PHE浓度较高,若回用于土壤修复,增加的PAH较多,降低了土壤洗涤的污染物去除率,故也不宜用于土壤修复;而从TX100达到吸附饱和到PHE达到穿透的这段时间内,虽然溶液中残留PHE,但仍可用于土壤修复,因为此时回用增加的PAH量小于10%的原始浓度,对污染物去除率的影响较小。为了达到回用表面活性剂同时保证污染物去除效果的目的,要求溶液在残留90%的表面活性剂的同时去除90%的PAH,并以此定义了表面活性剂的有效回收时间(∆t,min)。对于单一PAH体系,该时间即为PHE穿透10%的时间(t0.1PHE,min)与TX100穿透90%的时间(t0.9TX100,min)之差(见式(1))。显然,该时间越长,越有利于表面活性剂的回收再利用。

不同条件下TX100的有效回收时间见表1。由表1可见:流量较低时,有效回收时间较长;活性炭填充量过少时,吸附柱长度过短,PHE的穿透时间要短于TX100的饱和时间,导致有效回收时间为负数。但柱子也不宜过长,因为长柱子可能会导致TX100的过量吸附,从而长时间达不到回用90%表面活性剂的效果。由表1还可见,对于同一浓度TX100而言,PHE浓度的变化未对TX100到达穿透点的时间产生明显影响。在实际工程中,这种行为对于回收是有利的,溶液中不同污染物有不同的浓度,但这些差异不会影响到TX100的回收。另一方面,对于PHE而言,TX100浓度变化对PHE到达穿透点的时间也影响不大,但浓度高的TX100会先到达穿透点,从而导致较大的∆t值。总之,在柱长固定的情况下,低流量、高浓度TX100和低浓度PHE的情况有利于表面活性剂的回收。
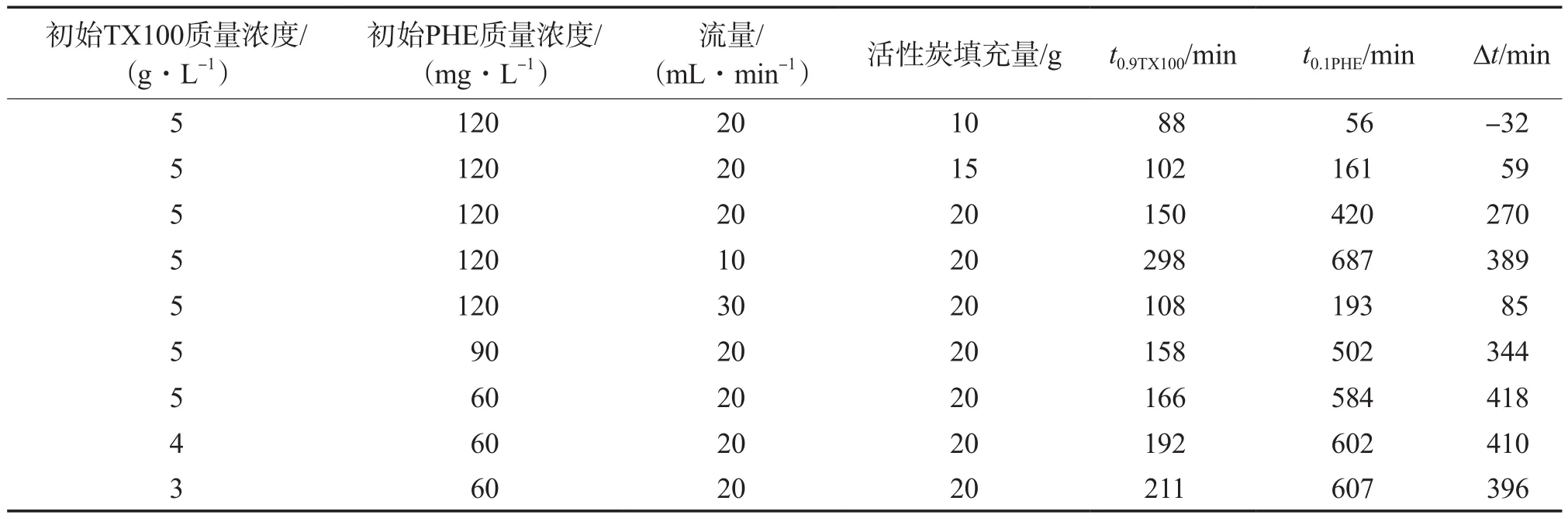
表1 不同条件下TX100的有效回收时间
2.4 模型拟合
2.4.1 Tomas模型
Tomas模型假设溶液呈活塞流形态,吸附符合Langmuir模型和二级反应动力学,且表面扩散是一个无限的扩散过程,是在固定床吸附中使用极为广泛的一种模型[27]。该模型的线性表达式为:
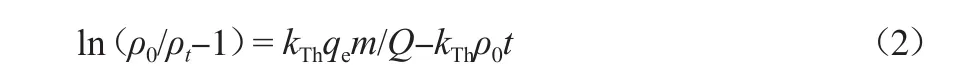
式中:kTh为Thoma模型常数,mL/(min·mg);qe为平衡吸附量,mg/g;m为活性炭填充量,g;Q为混合溶液的流量,mL/min。
Tomas模型的拟合结果见表2。由表2可见:该模型对于TX100和PHE的模拟效果均较好;对于TX100而言,高活性炭填充量、高流量和低浓度的kTh较高,而低活性炭填充量、低流量和高浓度时的单位吸附能力提高。这可能是由于吸附过程是由浓度差驱动的,即表面扩散的推动力是吸附量梯度,而细孔作用力与物理吸附较弱。通过吸附模型分析可以看出,PHE和TX100在固定床吸附中有相似的规律,即:随着活性炭填充量的减小平衡吸附量提高,这是因为对于相同的溶液,少量的活性炭有更多的吸附机会,从而提高了单位吸附量;随着流量的降低,吸附时间延长,从而增加了吸附量;PHE吸附量随着其浓度的降低而减小,其原因是PHE浓度较低时可提供的吸附物质有限。TX100吸附量基本不受PHE浓度变化的影响,这说明污染物浓度的高低未对表面活性剂的回收产生影响。
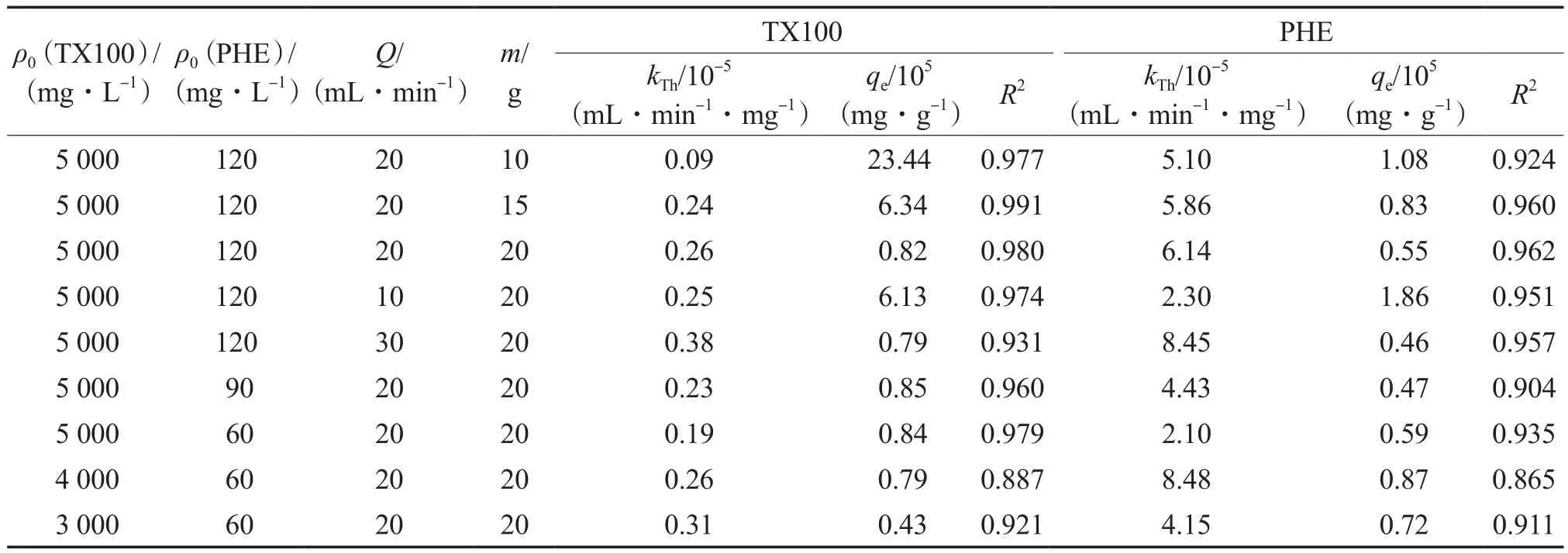
表2 Tomas模型的拟合结果
2.4.2 BDST模型
BDST模型是一种简化的关于吸附柱高度和穿透时间之间的关系,假设吸附是受吸附质和吸附剂的表面反应所控制[28],其表达式为:

式中:N0为固定床吸附容量,mg/L;Z为活性炭高度,cm;u为吸附质下降速率,cm/min;ka为吸附速率系数,L/(mg·min)。
由式(3)可以看出,在一定的初始浓度和流量以及一定的穿透浓度条件下,固定床的床高和穿透时间呈线性关系。在TX100穿透达到90%和10%以及PHE穿透达到10%时,BDST模型的线性表达式分别见式(4)~(6)。

从直线斜率可以看出,对于TX100的不同穿透时间和深度的两条直线几乎是平行的,这两条直线的水平距离称为交换区。由于PHE的浓度低,故需要更多的穿透时间,虽然实验没有得到PHE的穿透时间,但可以预期,其交换区要远大于TX100,也就是说当TX100穿透达到90%时,虽然活性炭对于TX100的吸附能力已经接近饱和,但仍有吸附PHE的能力。因此,流出液会保持一段相当长的时间可实现回用TX100,直到PHE的穿透达到90%。由于TX100和PHE的性质差异,其在吸附过程中的控制参数不同,从而导致其吸附过程中有不同的吸附速率,这为二者的分离提供了条件。
2.5 固定床节约运行成本分析
活性炭的吸附容量可以用下式[29]计算:

式中:qcolumn为活性炭固定床的吸附容量,g/g;ρ0.9和ρ0.1分别为流出液达到原浓度90%和10%时的质量浓度,mg/L;t0.9和t0.1分别为流出液质量浓度达到ρ0.9和ρ0.1时所需的时间,min。
以去除90%的PAH的同时回收90%的表面活性剂作为固定床分离的标准,计算可得每处理10 L废水需用活性炭234 g。考虑活性炭的再利用,以每kg活性炭1.3元计算,则活性炭的成本为0.3元。吸附后可以回收10 L浓度为3 g/L的表面活性剂溶液,即30 g表面活性剂,以表面活性剂30元/kg计,则为0.9元,即可以节省成本0.6元(0.06元/L)。固定床回收工艺增加了活性炭的购买费用,减少了表面活性剂的使用费用,总药剂成本减少。在实际工程中污染物质浓度要比实验中所采用的浓度要低,因此回收时间还可以延长,即节约的成本还可以增加。
3 结论
a)流量越低、活性炭填充量越多、表面活性剂浓度越高、PAH浓度越低,越有利于表面活性剂的回收和PAH的去除。
b)定义了有效回收时间,该时间是指PAH穿透10%的时间与表面活性剂穿透90%的时间之差。该时间越长,越有利于表面活性剂的回收。
c)BDST和Thomas模型对TX100和PHE吸附的模拟效果均较好。BDST模型拟合结果表明,当活性炭对TX100的吸附接近饱和时,仍对PHE有较强的吸附能力。
d)以去除90%PAH的同时回收90%表面活性剂为处理目标,考虑活性炭的再生,则活性炭固定床吸附分离法可节约运行成本0.06元/L。
[1] Wang Wentao,Massey Simonich S L,Xue Miao,et al.Concentrations,sources and spatial distribution of polycyclic aromatic hydrocarbons in soils from Beijing,Tianjin and surrounding areas,North China[J]. Environ Pollut,2010,158(5):1245 - 1251.
[2] Baek S O,Field R A,Goldstone M E,et al. A review of atmospheric polycyclic aromatic hydrocarbons:Sources,fate and behavior[J]. Water,Air,Soil Pollut,1991,60(3):279 - 300.
[3] Xia Zhonghuan,Duan Xiaoli,Qiu Weixun,et al.Health risk assessment on dietary exposure to polycyclic aromatic hydrocarbons(PAHs)in Taiyuan,China[J].Sci Total Environ,2010,408(22):5331 - 5337
[4] Wilson D J,Clarke A N. Hazardous waste site soil remediation:Theory and application of innovative technologies[J]. J Environ Qual,1995,24(2):384.
[5] Paria S. Surfactant-enhanced remediation of organic contaminated soil and water[J]. Adv Colloid Interface Sci,2008,138(1):24 - 58.
[6] Laha S,Tansel B,Ussawarujikulchai A. Surfactantsoil interactions during surfactant-amended remediation of contaminated soils by hydrophobic organic compounds:A review[J]. J Environ Manage,2009,90(1):95 - 100.
[7] Bennett G F. Reuse of surfactants and cosolvents for NAPL remediation[J]. J Hazard Mater,2002,89(1):99 - 101.
[8] Jiang Jianshen,Vane L M,Sikdar S K. Recovery of VOCs from surfactant solutions by pervaporation[J]. J Membr Sci,1997,136(1/2):233 - 247.
[9] Kungsanant S,Kitiyanan B,Rirksomboon T,et al.Recovery of nonionic surfactant from VOC-contaminated coacervate phase solutions by co-current vacuum stripping:Effect of surfactant concentration,temperature,and solute type[J]. Sep Purif Technol,2009,66(3):510 - 516.
[10] Cheng Hefa,Hu Yuanan,Luo Jian,et al. Multipass membrane air-stripping(MAS)for removing volatile organic compounds(VOCs)from surfactant micellar solutions[J]. J Hazard Mater,2009,170(2/3):1070 - 1078.
[11] Lee Dal-Heui,Cody R D,Kim Dong-Ju. Surfactant recycling by solvent extraction in surfactant-aided remediation[J]. Sep Purif Technol,2002,27(1):77 - 82.
[12] Vanjara A K,Dixit S G. Recovery of cationic surfactant by using precipitation method[J]. Sep Technol,1996,6(1):91 - 93.
[13] An Youn-Joo. Photochemical treatment of a mixed PAH/surfactant solution for surfactant recovery and reuse[J].Environ Prog Sustain,2001,20(4):240 - 246.
[14] Kowalska I. Surfactant removal from water solutions by means of ultrafiltration and ion-exchange[J]. Desalination,2008,221(1/3):351 - 357.
[15] Chiu Chun-Yu,Chen Yi-Hung,Huang Yi-Haw. Removal of naphthalene in Brij 30-containing solution by ozonation using rotating packed bed[J]. J Hazard Mater,2007,147(3):732 - 737.
[16] Doulia D,Xiarchos I. Ultrafiltration of micellar solutions of nonionic surfactants with or without alachlor pesticide[J]. J Membr Sci,2007,296(1/2):58 -64.
[17] Purkait M K,DasGupta S,De S. Removal of dye from wastewater using micellar-enhanced ultrafiltration and recovery of surfactant[J]. Sep Purif Technol,2004,37(1):81 - 92.
[18] Cheng Hefa,Sabatini D A. Separation of organic compounds from surfactant solutions:A review[J]. Sep Purif Technol,2007,42(3):453 - 475.
[19] Ahn Chi K,Kim Young M,Woo Seung H,et al.Selective adsorption of phenanthrene dissolved in surfactant solution using activated carbon[J]. Chemosphere,2007,69(11):1681 - 1688.
[20] Li Helian,Qu Ronghui,Li Chao,et al. Selective removal of polycyclic aromatic hydrocarbons(PAHs)from soil washing effluents using biochars produced at different pyrolytic temperatures[J]. Bioresour Technol,2014,163:193 - 198.
[21] 刘剑飞,张新. 活性炭对非离子型表面活性剂中菲的吸附能效[J]. 环境科学研究,2015,28(9):1481 - 1486.
[22] 李焱,赵纪光,凡明,等. 固定床活性炭干法烟气脱硫过程的模拟研究[J]. 化工环保,2016,36(3):317 - 320.
[23] Yang Jung-Seok,Baek K,Kwon Tae-Soon,et al.Adsorption of chlorinated solvents in nonionic surfactant solutions with activated carbon in a fixed bed[J]. J Ind Eng Chem,2009,15(6):777 - 779.
[24] Chen Nan,Zhang Zhenya,Feng Chuanping,et al.Investigations on the batch and fixed-bed column performance of fluoride adsorption by Kanuma mud[J].Desalination. 2011,268(1/3):76 - 82.
[25] Chen Suhong,Yue Qinyan,Gao Baoyu,et al. Adsorption of hexavalent chromium from aqueous solution by modified corn stalk:A fixed-bed column study[J]. Bioresour Technol,2012,113:114 - 120.
[26] Ahmad A A,Hameed B H. Fixed-bed adsorption of reactive azo dye onto granular activated carbon prepared from waste[J]. J Hazard Mater,2010,175(1/3):298 - 303.
[27] Bohart G S,Adams E Q. Some aspects of the behavior of charcoal with respect to chlorine[J]. J Am Chem Soc,1920,42(3):523 - 544.
[28] Cunha G D C,Romão L P C,Santos M C,et al. Adsorption of trihalomethanes by humin:Batch and fixed bed column studies[J]. Bioresour Technol,2010,101(10):3345 - 3354.
[29] 韩晓琳,邱兆富,胡娟,等. 水处理活性炭吸附容量指标测定方法的优化及应用[J]. 环境污染与防治,2013,35(1):54 - 59.
