分子动力学模拟在复合固体推进剂研究中的应用
2018-01-04苗瑞珍高晓敏乔小平陈海洋郗明颇
苗瑞珍,高晓敏,乔小平,陈海洋,郗明颇,孟 勇
(西安北方惠安化学工业有限公司, 西安 710302)
【弹药工程】
分子动力学模拟在复合固体推进剂研究中的应用
苗瑞珍,高晓敏,乔小平,陈海洋,郗明颇,孟 勇
(西安北方惠安化学工业有限公司, 西安 710302)
概述了分子动力学模拟方法在复合固体推进剂的组分相容性、感度及安全性研究中的应用和分子动力学在研究固体推进剂中炸药爆轰及燃烧反应机理方面的应用,为从分子层面解释炸药爆炸时的微观反应机理提供了可能;分析了目前存在的问题及发展前景,可为相关科研人员提供理论参考。
分子动力学模拟;固体推进剂;性能;反应机理
1 前言
复合固体推进剂是一种以高分子粘合剂为基体,添加氧化剂、增塑剂和燃烧剂等填料制成的一种推进剂[1],随着化学、材料等学科的不断发展以及火箭技术的不断进步,对固体推进剂的性能要求越来越高,高能、钝感和低特征信号成为复合固体推进剂发展的主要目标[2]。性能优异的复合固体推进剂配方的研制,需要耗费大量的人力、物力和财力,不仅研制周期较长,同时实验还具有一定的危险性。1993年,Cumming等[3]在25届ICT国际会议上报道了HMX和PNMO相互作用的分子力学(MM)和分子动力学(MD)模拟计算工作,开创了近代计算化学对含能材料分子间相互作用理论研究的先河。美国伊利诺伊大学先进火箭发动机仿真中心(CSAR)自1997年开始就在美国能源部ASCI计划的资助下进行了固体火箭发动机全系统仿真[4],其中一部分就涉及到复合固体推进剂的模拟研究[5]。近年来,随着理论化学和计算化学以及计算机模拟技术的迅速发展,越来越多的研究者开始应用计算机模拟技术对固体推进剂的结构、性能和微观反应机理进行研究以弥补实验手段和理论研究在这方面的一些不足之处,以期为固体推进剂的配方设计,新材料设计、开发提供前期的理论预测与科学依据。
本文主要介绍了MD模拟方法在复合固体推进剂的性能及其炸药微观反应机理研究方面的应用进展,分析应用中存在的问题并展望未来的发展前景,以期为科研人员利用该方法开展固体推进剂研究提供参考。
2 在复合固体推进剂性能研究中的应用
2.1 在复合固体推进剂相容性研究中的应用
为了改善固体推进剂的低温力学性能,降低其玻璃化转变温度,同时增加其柔韧性并优化加工性能,需要在配方中加入功能组分增塑剂,理想的增塑剂必须与粘结剂具有良好的相容性,才能保证推进剂具有优异的综合性能[6-9]。此外也可以在炸药中加入一定量相容性较好的高聚物粘结剂来改善其力学性能及加工性能。
根据高分子溶液理论,由粘合剂和增塑剂组成的混合体系可以视为高分子溶液,一般情况下溶解是溶质分子和溶剂分子互相混合作用的过程,两种物质混合时,在恒温恒压下,这种过程能自发进行,形成相容体系的热力学条件是:
ΔGM= ΔHM-TΔSM<0
(1)
其中:T是溶解时温度;ΔSM、ΔGM和ΔHM分别代表混合熵,混合自由能和混合焓。
对于高分子体系, 如果异种分子间没有相互作用(如氢键), 那么ΔHM值总是大于零的, 因此,混合焓项始终不利于两者的混合, 能否均匀混合取决于熵项ΔSM的贡献是否能克服混合焓项。实际中, 对于推进剂内高分子材料在混合过程中, 熵的增加非常小[10],因此ΔGM值的正负取决于ΔHM的大小。 Hildebrand等[11]通过研究表明物质间的相互作用能力决定于其内聚能密度, 因此引入了溶度参数的概念, 其定义为内聚能密度的平方根:
δ=(ΔE/V)1/2=[(ΔHV-RT)/V]1/2
(2)
ΔE、V、ΔHV分别为体系的内能、体积和蒸发热。高分子材料混合过程中的焓变与高分子的溶度参数的关系为[20]:
ΔHM/V=(δ1-δ2)2φ1φ2
(3)
φ1、φ2分别为组分1、2 的体积分数. 由(3)式可见, 组分1、2 的溶度参数δ1、δ2越接近, ΔHM值越小, 体系相容性越好。因此, 溶度参数差值(Δδ)可以作为组分间相容性理论预测指标。
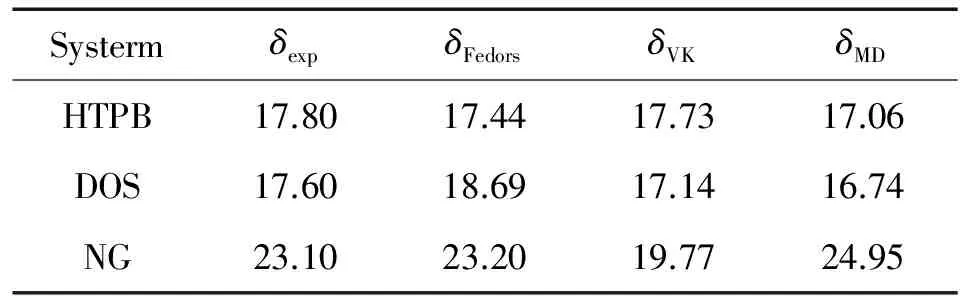
表1 采用不同方法所得溶度参数δ (J1/2·cm-3/2)[12]

表2 采用不同方法计算的Δδ(J1/2·cm-3/2) [12]
付一政等[12]用MD模拟方法预测了聚丁二烯(HTPB)和增塑剂葵二酸二辛酯(DOS)以及硝化甘油(NG)之间的相容性。表1为用MD模拟计算所得纯物质的溶度参数和实验值[13-15], 以及理论值(目前现有的从理论上计算高分子溶度参数的主要方法是Dunkel[16]发展的原子与基团贡献法。表1同时给出了根据原子与基团贡献法原理采用不同估算方法计算的值,其中δFedors、δvanKrevelen分别为采用Fedors[17]和van Krevelen[18]参数求得的溶度参数)。由表可得, 对于HTPB、DOS、NG 来说δFedors、δVK和δMD均与实验值吻合得较好, 说明通过MD 模拟可以得到与实验值比较吻合的溶度参数。
表2给了采用不同方法所得的Δδ值,有研究表明[19]对于分子间不存在强作用(如极性集团或者氢键作用)的高分子体系,两种材料的Δδ只要满足∣Δδ∣<(1.3-2.1) J1/2·cm-3/2, 表明两者就可以相容。综合表1 和2,不同的计算方法得到不同的溶度参数值,但是通过计算Δδ判断共混物相容性的结果均一致, 即∣Δδ∣HTPB/DOS<(1.3-2.1) J1/2·cm-3/2, HTPB/DOS 共混物属于相容体系, 而∣Δδ∣HTPB/NG的值较大,与相容性较好的HTPB/DOS体系相比, HTPB与NG的相容性较差,属于不相容体系, 这一结论与实验结果一致,由此说明MD模拟可以作为研究推进剂相容性的可靠方法。
李倩[20-21]则对高能推进剂中叠氮粘合剂与硝酸酯进行了 MD 模拟,得出了与实际相符的各组分的溶度参数;Li 等[22]从介观尺度计算了 NEPE 推进剂粘合剂与增塑剂间的相分离情况;Abou-Rachid采用MD方法研究了固体推进剂的相容性[23];许晓娟,肖继军等[24]运用MD方法分别预测了以下5种体系相容性和稳定性大小的顺序依次为:ε-CL-20/PEG>ε-CL-20/Estane5703>ε-CL-20/GAP>ε-CL-20/HTPB>ε-CL-20/F2314,与实验结果一致;焦东明等[25]运用MD方法计算了HTPB粘合剂及增塑剂DOS、DOA、TOA、DBP和DOP的溶度参数,并以此来选择合适的增塑剂,计算数值基本吻合实验值;王欢等[26]运用MD方法研究了固体推进剂BTTN、NG、HMX、HTPB、DOS,EC等的相容性,结果表明,BTTN、NG均与EC相容,HTPB与DOS是相容体系,而HTPB/NG、HMX/BTTN和HMX/NG体系均为不相容体系。这些模拟计算为固体推进剂的配方设计与发展提供了强有力的理论指导和技术支持。
2.2 在复合固体推进剂感度及安全性研究中的应用
通常情况下分子中引发键的键级越小,键长越大,即越容易断裂,引发分解和起爆,相应的其感度就越高。经典模拟过程不涉及键级,但可以给出键长的统计平均值。肖鹤鸣课题组以及国外的其他研究中曾使用引发键最大键长(Lmax)研究了含能材料的感度,所得结论相符实验数据,说明釆用随来关联含能材料的感度是可靠的。
此外力学性能密切关联感度,若推进剂体系的力学强度较小,就使得其刚性较小,柔性增强,即体系变“软”,在体系受到外力作用时,可以有效缓冲和分散外力作用,减小炸药颗粒之间的摩擦,使其内部应力分布均匀,从而减少“热点”的形成,减小其感度,增加安全性。
结合能也是含能材料稳定性及安全性的一个重要参考指标,结合能(Ebind)为相互作用能(Einter)的负值,对于某一个两组分体系A与B而言,A与B的之间的相互作用能等于平衡结构的总能量(Etotal)减去除掉A组分后该结构的能量(EB),再减去除掉B组分后该结构的能量(EA),即:
Ebind=-Einter=-(Etotal-EA-EB)
结合能是组分相互作用强弱的标志。结合能越大,表示组分间的相互作用越强,形成的体系越稳定,相容性越好,对结合能的贡献主要是非键相互作用引起的,其中静电作用力所占比例最大,范德华能量项所占比例最小。各组分间若存在强烈的分子间作用力,会增加了炸药分子体系的力学稳定性以及热力学稳定性,提高了共晶分子对机械外力的抗振性与耐热性,也即降低了其撞击感度以及热感度,从而提高其安全性。
刘强等[27]运用MD方法计算了CL-20/TNT共晶不同晶面以及RDX/TNT共混体系的最大引发键键长,力学性能以及结合能与感度和其体系安全性的关系,由计算结果可知CL-20/TNT共晶体系中(100)晶面Lmax最大,拉伸模量、剪切模量和体积模量的排序依次为(3×2×1)>(001)>(010)>(100)>(120),而对于RDX/TNT共混体系来说,混合体系中RDX组分的引发键(N-O2)的Lmax均比纯的RDX晶体短,说明TNT的加入会使体系感度下降,随着体系中TNT组分含量的增加,其拉伸模量和剪切模量逐渐增大,体系刚性增加,感度增大,此结论与实验结果能够很好的吻合;肖鹤鸣课题组近年来采用MD方法模拟了多种含能材料及其复合物的结构性能与感度的关系并取得一定进展,丰富了高能复合物配方研制理论体系;付一政等人[28]采用MD模拟方法对CL-20、DNB、两者的共混物及共晶的感度、结合能和力学性能进行了模拟计算,结果表明共晶和共混均会降低体系的感度,但共晶效果更加明显;与共混物结构相比,共晶结构更加稳定,共晶和共混均可以改变复合体系的力学性能,降低体系的刚度,增加体系的柔性和安全性,但共混会使体系的力学性能劣化,与实验结果一致。感度是衡量推进剂安全性的重要参数,MD模拟为感度研究提供了一种更简单有效的方法。
2.3 在复合固体推进剂爆轰及燃烧反应机理中的应用
基于反应力场的MD模拟方法是连接量子化学和分子动力学方法的桥梁,为大规模研究凝聚态性质及处理其中可能发生的化学反应过程提供了可能。近几年反应力场在材料科学尤其对于固体推进剂中炸药的爆轰及燃烧反应的研究中的应用越来越广泛,不仅可以对新型炸药的设计和使用过程提供理论依据,而且可以对分子原子级别的微观物理化学现象进行解释。其中用于研究反应机理的力场现在运用最多的是ReaxFF力场,它已经成功用于一些固体推进剂中炸药的爆轰及燃烧反应的机理研究中。如三硝基甲苯(TNT),环三亚甲基三硝铵(RDX),季戊四醇四硝酸酯 (PETN),环四亚甲基四硝铵(HMX),1,3,5三胺基-2,4,5-三硝基苯(TATB),三过氧化三丙酮(TATP),硝基甲烷(NM)的冲击起爆、冲击点火和爆轰反应等,这不仅对含能材料的理论设计进行指导,而且对材料的使用、存储、运输安全问题都有重要的现实意义。
硝基甲烷其实并不是实际应用的炸药,但是由于含有NO2基团,可以代表一类含有硝基的炸药,鉴于其简单的分子结构,且在冲击作用下可以发生爆轰现象所以被广泛用于炸药反应的模拟中。Naomi Rom等[29]研究了压力对硝基甲烷分解路径的影响,发现在高压和低压下热解引发反应存在一个竞争。Han等[30]研究了高压下固相硝基甲烷的分解机理,发现在较低的温度2000K和2500K时单个硝基甲烷分子会发生分子内的重排,生成亚硝酸酯(CH3ONO),接着发生C-N键的断裂,而在较高的温度3000K时两个硝基甲烷分子间会发生质子转移形成C3HNOOH和 CH2NO2,说明温度会对反应路径有较大的影响。Strachan等[31]研究了密度对环三亚甲基三硝胺(RDX)晶体热解过程的影响,得到了随着密度的变化各产物数量的变化规律以及低密度与高密度晶体分解机理的主要区别。Strachan等[32]研究了不同冲击速度下RDX的分解反应,结果表明冲击速度对RDX热解反应机理有较大的影响。Michael F. Russo等[33]研究了不同当量比的1,5-二硝基缩二脲(DNB)与硝酸的燃烧反应,研究表明在一定的当量比时,会发生自燃反应,释放出大量的热能。M.R. Weismiller等[34]运用ReaxFF力场对硼烷氨氧化反应的动力学机理进行了研究。Adri C. T. van Duin等[35]研究了硼烷氨的脱氢反应和燃烧反应的机理,文中首先是运用量子力学计算得到的参数对硼烷氨的力场参数进行拟合,然后对硼烷氨单分子和多分子的热解和燃烧反应进行了分子动力学模拟,得到产物第一分子H2的生成是分子内部的反应,反应的活化能是26.36kcal/mol,这与实验值117 kJ/mol很接近[36]。这些模拟计算为固体推进剂的配方设计与发展提供了强有力的理论指导和技术支持。
3 结论
通过对已有的火炸药品种进行分子动力学模拟研究,不仅可以弥补宏观实验手段研究尺度的不足,而且有助于认识火炸药结构与性能之间的内在关系和作用机理,丰富火炸药基础理论体系,为其配方设计和工艺参数的选择提供参考。
采用MD模拟方法对新型火炸药品种的进行配方设计和性能预测,能够大大缩短研制周期,降低成本,提高研发过程中的安全性。
由于一般推进剂中火炸药的反应条件比较极端,反应速度非常快,用常规的实验手段无法捕捉一些微观信息,目前对火炸药的分子动力学研究大部分仅仅是模拟仿真,缺少实验验证。由于计算机性能及模拟效率等因素的限制,模拟体系选取的原子数量有限,模拟规模较小,以及目前力场的不完善,仿真模型与实际情况存在较大的差距。
MD模拟方法在目前的应用中存在一定的局限性,但通过理论与实验研究紧密结合,MD模拟方法必将成为研制高性能复合固体推进不可或缺的研究手段。
[1] 任务正, 王泽山.火炸药理论与实践 [M].北京: 中国北方化学工业总公司, 2001: 120-135.
[2] 张晓宏, 莫红军.下一代战术导弹固体推进剂研究进展 [J].火炸药学报, 2007, 30(1): 24-27.
[3] CUMMING A, LEIPER G, ROBSON E.Molecular modelling as a tool to aid the design of polymer bonded explosives [J].Journal of Hazardous Materials 1993, 3: 23-26.
[4] DICKFL W A, FIEDLER R A, HEATH M T.Integrated simulation of solid propellant rockets [J].2000, 11: 21-24.
[5] DICK W A, HEATH M T, FIEDLER R A, et al.Advanced Simulation of Solid Propellant Rockets from First Principles [J].Journal of Hazardous Materials, 2005, 4: 10-13.
[6] EDGAR K J, BUCHANAN C M, DEBENHAM J S, et al.Advances in cellulose ester performance and application[J].Progress in Polymer Science, 2001, 26(9):1605-1688.
[7] MUTHIAH R, SOMASUNDARAN U I, VERGHESE T L, et al.Energetics and Compatibility of Plasticizers in Composite Solid Propellants[J].Defence Science Journal, 2013, 39(2).
[8] MATHIEU J, STUCKI H.Military high explosives [J].CHIMIA International Journal for Chemistry, 2004, 58(6): 383-389.
[9] JAWALKAR S N, MEHILAL, RAMESH K, et al.Studies on the effect of plasticiser and addition of toluene diisocyanate at different temperatures in composite propellant formulations.[J].Journal of Hazardous Materials, 2009, 164(2):549-554.
[10] SUN X Q, FAN X W, XUE-HAI J U, et al.Research Methods on Component Compatibility of Propellants[J].Chemical Propellants & Polymeric Materials, 2007, 152(4), 362-369.
[11] HILDEBRAND J H,SCOTT R L.The solubility of non-electrodytes[M].NewYork: Reinhold Publishing Corp., 1950: 424-434.
[12] 付一政, 刘亚青, 兰艳花.端羟基聚丁二烯/增塑剂共混物相容性的分子动力学模拟[J].物理化学学报, 2009, 25(7):1267-1272.
[13] 杨月诚, 焦东明, 强洪夫,等.HTPB推进剂组分溶度参数的分子模拟研究[J].含能材料, 2008, 16(2):191-195.
[14] ABOU-RACHID H, LUSSIER L S, RINGUETTE S, et al.On the Correlation between Miscibility and Solubility Properties of Energetic Plasticizers/Polymer Blends: Modeling and Simulation Studies[J].Propellants Explosives Pyrotechnics, 2008, 33(4):301-310.
[15] 任玉立, 陈少镇.关于硝化纖維素濃溶液的研究——体系溶度参数与相溶性[J].火炸药, 1981(1):11-18.
[16] DUNKEL M.Calculation of intermolecular forces in organic compounds [J].PhysChem, 1928, 138: 42-54.
[17] FEDORS R F.A method for estimating both the solubility parameters and molar volumes of liquids [J].Polymer Engineering & Science, 1974, 14(2): 147-154.
[18] KREVELEN D W.Properties of polymers,3rd Edition [M].Elsevier Science Pub Co, 1990: 188-204.
[19] MANSON J A, SPERLING L H.Polymer Blends and Composites[M].Heyden, 1976.
[20] 李倩, 姚维尚, 谭惠民.叠氮粘合剂与硝酸酯溶度参数的分子动力学模拟[J].含能材料, 2007, 15(4):370-373.
[21] 李倩, 姚维尚, 谭惠民.分子动力学模拟叠氮热塑性弹性体的杨氏模量及其与硝酸酯的溶度参数[J].火炸药学报, 2007, 30(4):13-16.
[22] LI S, LIU Y, TUO X, et al.Mesoscale dynamic simulation on phase separation between plasticizer and binder in NEPE propellants[J].Polymer, 2008, 49(11):2775-2780.
[23] ABOU-RACHID H, LUSSIER L, RINGUETTE S, et al.On the correlation between miscibility and solubility properties of energetic plasticizers/polymer blends: modeling and simulation studies [J].Propellants, Explosives, Pyrotechnics, 2008, 33(4): 301-310.
[24] 许晓娟, 肖继军, 黄辉,等.ε-CL-20基PBX结构和性能的分子动力学模拟HEDM理论配方设计初探[J].中国科学:, 2007, 37(6):556-563.
[25] 焦东明, 杨月诚, 强洪夫,等.HTPB固体推进剂增塑剂选取分子模拟研究[J].化学研究与应用, 2009, 21(6):805-809.
[26] 王欢, 孙治丹, 张常山, 等.固体推进剂组分相容性的分子动力学模拟[J].火炸药学报, 2016, 39(5): 69-73.
[27] 刘强.RDX 与 CL-20 及其共晶和复合体系的 MD 模拟研究[D].南京: 南京理工大学, 2014.
[28] 付一政, 康志鹏, 郭志婧, 等.共晶和共混对 CL-20/DNB 感度和热解机理影响的 MD 模拟[J].含能材料, 2017, 25(2): 94-99.
[29] ROM N, ZYBIN S V, VAN DUIN A C T, et al.Density-Dependent Liquid Nitromethane Decomposition: Molecular Dynamics Simulations Based on ReaxFF[J].The Journal of Physical Chemistry A, 2011, 115(36): 10181-10202.
[30] LIU J, CHAMBREAU S D, VAGHJIANI G L.Thermal Decomposition of 1, 5-Dinitrobiuret (DNB): Direct Dynamics Trajectory Simulations and Statistical Modeling[J].The Journal of Physical Chemistry A, 2011, 115(28): 8064-8072.
[31] STRACHAN A, KOBER E M, VAN DUIN A C, et al.Thermal decomposition of RDX from reactive molecular dynamics[R].California Inst of Tech Pasadena Materials and Processes Simulation Center, 2005.
[32] STRACHAN A, VAN DUIN A C T, CHAKRABORTY D, et al.Shock waves in high-energy materials: The initial chemical events in nitramineRDX[J].Physical Review Letters, 2003, 91(9): 098301.
[33] RUSSO JR M F, BEDROV D, SINGHAI S, et al.Combustion of 1, 5-Dinitrobiuret (DNB) in the Presence of Nitric Acid Using ReaxFF Molecular Dynamics Simulations[J].The Journal of Physical Chemistry A, 2013, 117(38): 9216-9223.
[34] WEISMILLER M R, RUSSO JR M F, VAN DUIN A C T, et al.Using molecular dynamics simulations with a ReaxFF reactive force field to develop a kinetic mechanism for ammonia borane oxidation[J].Proceedings of the Combustion Institute, 2013, 34(2): 3489-3497.
[35] WEISMILLER M R, DUIN A C T, LEE J, et al.ReaxFF reactive force field development and applications for molecular dynamics simulations of ammonia borane dehydrogenation and combustion[J].The Journal of Physical Chemistry A, 2010, 114(17): 5485-5492.
[36] BROWN C M, JACQUES T L, HESS N J, et al.Dynamics of ammonia borane using neutron scattering[J].Physica B: Condensed Matter, 2006, 385: 266-268.
ApplicationofMolecularDynamicsMethodinResearchofSolidPropellants
MIAO Ruizhen, GAO Xiaomin, QIAO Xiaoping, CHEN Haiyang, XI Mingpo, MENG Yong
(Xi’an North Huian Chemical Industry Co., Ltd., Xi’an 710302, China)
The application progress of molecular dynamics simulation method inresearch of component compatibility, sensitivity and safety of composite solid propellants at home and abroad is summarized. In addition, the application of molecular dynamics in the study of detonation and combustion reactions in solid propellants is also summarized.It is possible to explain the microscopic reaction mechanism of explosive explosion at the molecular level.And the existing problems in applications and development prospects are analyzed. The references are provided for scientific researchers in this field to develop the research work of propellants and explosives using this method.
molecular dynamics simulations; solid propellant;properties; reaction mechanism
2017-08-15;
2017-08-30
苗瑞珍(1990—),女,硕士,主要从事含能材料结构与性能研究。
10.11809/scbgxb2017.12.010
本文引用格式:苗瑞珍,高晓敏,乔小平,等.分子动力学模拟在复合固体推进剂研究中的应用[J].兵器装备工程学报,2017(12):40-44.
formatMIAO Ruizhen, GAO Xiaomin, QIAO Xiaoping, et al.Application of Molecular Dynamics Method in Research of Solid Propellants[J].Journal of Ordnance Equipment Engineering,2017(12):40-44.
TJ55
A
2096-2304(2017)12-0040-05
(责任编辑周江川)
