中药多糖及人血清中糖蛋白N-糖链的毛细管电泳指纹图谱研究
2017-12-27管月清张艳梅康经武
李 凤,管月清,张艳梅,康经武
(1.西安文理学院 化学工程学院,陕西 西安 710065;2.生命有机化学国家重点实验室,中国科学院上海有机化学研究所,上海 200032;3.上海市农业科学院农产品质量标准与检测技术研究所,上海 200032)
中药多糖及人血清中糖蛋白N-糖链的毛细管电泳指纹图谱研究
李 凤1*,管月清2,张艳梅3,康经武2
(1.西安文理学院 化学工程学院,陕西 西安 710065;2.生命有机化学国家重点实验室,中国科学院上海有机化学研究所,上海 200032;3.上海市农业科学院农产品质量标准与检测技术研究所,上海 200032)
该文建立了激光诱导荧光毛细管电泳(CE-LIF)检测中药多糖和人血清糖蛋白中N-糖链的分析方法。利用8-氨基芘-1,3,6-三磺酸钠(APTS)为柱前荧光衍生试剂,结合CE分析,可在15 min内同时测定7种中性单糖。该方法应用于中药制剂和药物植物中单糖组成和含量的测定,加标回收率为88.0%~102%,相对标准偏差(RSD)小于3.0%。该方法灵敏度高、准确性好、实用可靠,适合于监控中药制剂中糖类物质的种类和含量。同时,基于毛细管凝胶电泳方法(CGE)建立了人血清中糖蛋白的N-糖链指纹图谱。利用中性涂层聚乙烯醇(PVA)有效地抑制电渗流,增加分离时间,实现了糖链异构体的有效分离。对5 μL 人血清进行毛细管电泳分析,共分离得到17个组分。这为进一步研究复杂生物体系血清蛋白的糖基化变化提供了可能性。
激光诱导荧光毛细管电泳;中药多糖;N-糖基化修饰;糖蛋白
糖类化合物是能量的主要来源,同时也是重要的功能分子。其中,多糖和糖蛋白是糖类化合物常见的两种存在形式。研究证实,多糖特别是水溶性多糖具有重要的生理活性,如抗肿瘤、抗炎、降血糖以及免疫调节功能等,是中药的重要功效成分之一[1]。而在真核生物中,蛋白糖基化是一类重要的蛋白翻译后修饰形式。糖蛋白中糖链不仅影响蛋白质的结构和性质,而且在分子识别、信号转导以及免疫应答等生物过程中也发挥重要作用[2]。不论是中药多糖还是糖蛋白中的糖链,其结构特征如相对分子质量及其分布、单糖组成以及连接顺序等与活性密切相关[3-4]。因此,对糖类化合物的结构表征为中药的质量控制和糖蛋白的功能研究提供了重要线索。
由于糖类化合物结构的复杂性和多样性,其分离是分析化学研究的难点之一[5-6]。毛细管电泳技术(CE)具有分离效率高、样品消耗少等优点,在复杂糖链结构表征中具有很大的应用潜力[7]。Karger等[8]报道了基于CE和激光诱导荧光(LIF)检测的糖链检测方法,7 min内实现了免疫球蛋白G(IgG)中12种糖链异构体的高效分离,并将该方法成功应用于单克隆抗体类药物糖型表征,为抗体类药物的质量控制提供了简单、快速、灵敏的分析方法。近年来,胶束毛细管电泳(MEKC)、毛细管区带电泳(CZE)及毛细管凝胶电泳(CGE)等多种电泳模式也被用于多糖及单糖的分离检测中,分析对象主要集中于植物多糖、单克隆抗体类药物等[9-13]。王文波等[11]利用毛细管电泳对原研体CD20人鼠嵌合单抗和4个生物类似药的N-糖谱进行分析,发现在单抗的质量控制中,应重视N-糖链检测的重要性。但对于复杂生物体系中糖蛋白的糖类化合物的结构表征报道较少。Callewaert课题组利用ABI 3030 DNA测序仪,建立了血清N-糖链简单、快速的检测方法,并进一步结合微流控芯片技术,大大缩短了分析时间,已应用于肝癌患者血清糖蛋白的糖型分析[14-15]。
本文采用荧光衍生试剂8-氨基芘-1,3,6-三磺酸钠(8-Aminopyrene-1,3,6- trisulfonic acid,APTS)对糖类化合物进行柱前衍生,采用毛细管区带电泳(CZE),利用硼酸盐与单糖衍生物的鳌合作用,15 min内实现了7种中性单糖的高效分离。该方法成功应用于中药制剂和药物植物中单糖组成和含量的测定。同时采用毛细管凝胶电泳(CGE)建立了人血清中N-糖链的指纹图谱。本研究利用聚乙烯醇(PVA)中性涂层,有效地抑制了电渗流,增加了分离时间,实现了糖链异构体的高效分离。
1 实验部分
1.1 仪器与试剂
P/ACE MDQ CE系统配LIF检测器(美国Beckman Coulter公司),激发波长488 nm,发射波长520 nm。弹性石英毛细管柱50 μm i.d.,360 μm o.d.(Polymicro Technologies公司,USA)。Agilent 7820A气相色谱仪。聚乙烯醇(PVA)中性涂层毛细管柱(实验室自制)。8-氨基芘-1,3,6-三磺酸钠盐(APTS)购自Invitrogen公司(USA);葡萄糖(Glu)、半乳糖(Gal)、阿拉伯糖(Ara)、甘露糖(Man)、木糖(Xyl)、鼠李糖(Rha)、麦芽糖(Mal)、氰基硼氢化钠、荧光素钠(fluorescein sodium,FS)、三乙胺、乙腈、乙酸、无水乙醇(分析纯)、四氢呋喃(分析纯)、聚乙烯醇(PVA)、人血清均购自Sigma公司。肽N-糖苷酶(PNGase F)购自New England Lab。Glyko®APTS-(Maltodextrin Ladder) 购自美国Prozyme公司。硼砂(分析纯)、三羟甲基氨基甲烷(Tris,分析纯)、氢氧化钠(分析纯)购自上海实生细胞生物技术有限公司。中药注射液(热毒宁注射液)与纯化后的药用植物多糖样品(金银花多糖)均由江苏康缘药业股份有限公司提供。实验用水为18 MΩ·cm超纯水(Millipore超纯水系统,美国Millipore公司)。溶液使用前需用0.22 μm滤膜过滤。
1.2 样品前处理溶液的制备
精确称取药用植物多糖,水溶、涡旋,预配成质量浓度为2 mg/mL的溶液,13 400 r/min离心5 min后取上清液备用。准确移取中药注射液及药用植物多糖样品各1 mL,分别加入4 mol/L盐酸1 mL,涡旋混匀后,80 ℃下密封反应6 h。冷却至室温后,N2吹干,再加1 mL 超纯水溶解水解样品,4 ℃冰箱保存备用。
人血清样品用3 kDa 超滤管处理以去除低分子量组分。取人血清样品5 μL(蛋白质量浓度 10 mg/mL)按照NEB的试剂盒进行样品前处理:加入10 μL 2% SDS 在60 ℃中孵育10 min使蛋白变性,依次加入5 μL 4% NP-40以及PNGase F 2 μL,37 ℃酶解24 h。加入一定体积的预冷乙醇,使其终浓度为75%,混匀后,-20 ℃静置0.5 h,13 400 r/min离心15 min。取上清液冷冻干燥后,溶于5 μL超纯水中备用。
1.3 APTS衍生化
分别取葡萄糖、半乳糖、甘露糖、阿拉伯糖、木糖、鼠李糖、麦芽糖的标准品配成质量浓度均为1 mg/mL的标准溶液。准确称取0.79 mg APTS,溶于30 μL(4.2 mol/L)乙酸溶液中,配成5.0×10-2mol/L APTS的乙酸溶液;准确称取1.9 mg 氰基硼氢化钠,溶于60 μL四氢呋喃溶液中,配成0.5 mol/L氰基硼氢化钠的THF溶液。
准确配制浓度为1.0×10-7mol/L的荧光素钠标准溶液,作为内标。取水解后的样品溶液、酶切后的N-糖链及混合单糖各系列溶液5 μL,分别加入2 μL APTS的乙酸溶液与4 μL氰基硼氢化钠的THF溶液,混合均匀后,于55 ℃下密封反应2 h。对于含有唾液酸的糖链,37 ℃下过夜衍生。避光冷却后稀释至100 μL,备用。
1.4 PVA 涂层柱的制备
配制6% PVA溶液,超声脱气10 min,截取4 m×50 μm i.d.的毛细管柱。氮气作用下,向毛细管柱通入6% PVA水溶液,持续2 h。之后去除未与毛细管表面结合的PVA。最后将其置于气相色谱仪的恒温箱中,以5 ℃/min程序升温至145 ℃,保持5 h。
1.5 CE分离条件
中药多糖的CZE分离条件:裸毛细管柱40 cm(有效长度29.5 cm);新毛细管用0.1 mol/L NaOH溶液活化30 min,再用超纯水和背景电解质溶液分别冲洗2 min。分离电压:20 kV;压力进样:0.3 psi×5 s;柱温为25 ℃;背景电解质为30 mmol/L硼砂缓冲溶液(pH 10.03)。
血清N-糖链的CGE分离条件:PVA涂层毛细管柱50 cm(有效长度39.5 cm);分离电压:20 kV;压力进样:0.3 psi×5 s;柱温为25 ℃;背景电解质为25 mmol/L NH4Ac(pH 4.75),0.4% PEG 20 000。
2 结果与讨论
2.1 中药多糖毛细管区带电泳方法的建立
2.1.1CZE分离谱图所分离的7种中性单糖不具备强的紫外或荧光发色基团,给检测带来了一定困难。本文选用8-氨基芘-1,3,6-三磺酸钠(APTS)作为柱前衍生试剂,对荧光衍生后的单糖进行分析。图1为7种单糖混合标样的毛细管区带电泳图。在30 mmol/L硼砂缓冲溶液(pH 10.03)中,7种单糖标准品在较短时间(15 min)内实现了基线分离。实验选用荧光素钠作为内标,可对衍生后各单糖的峰面积进行校正,从而提高定量的准确性。
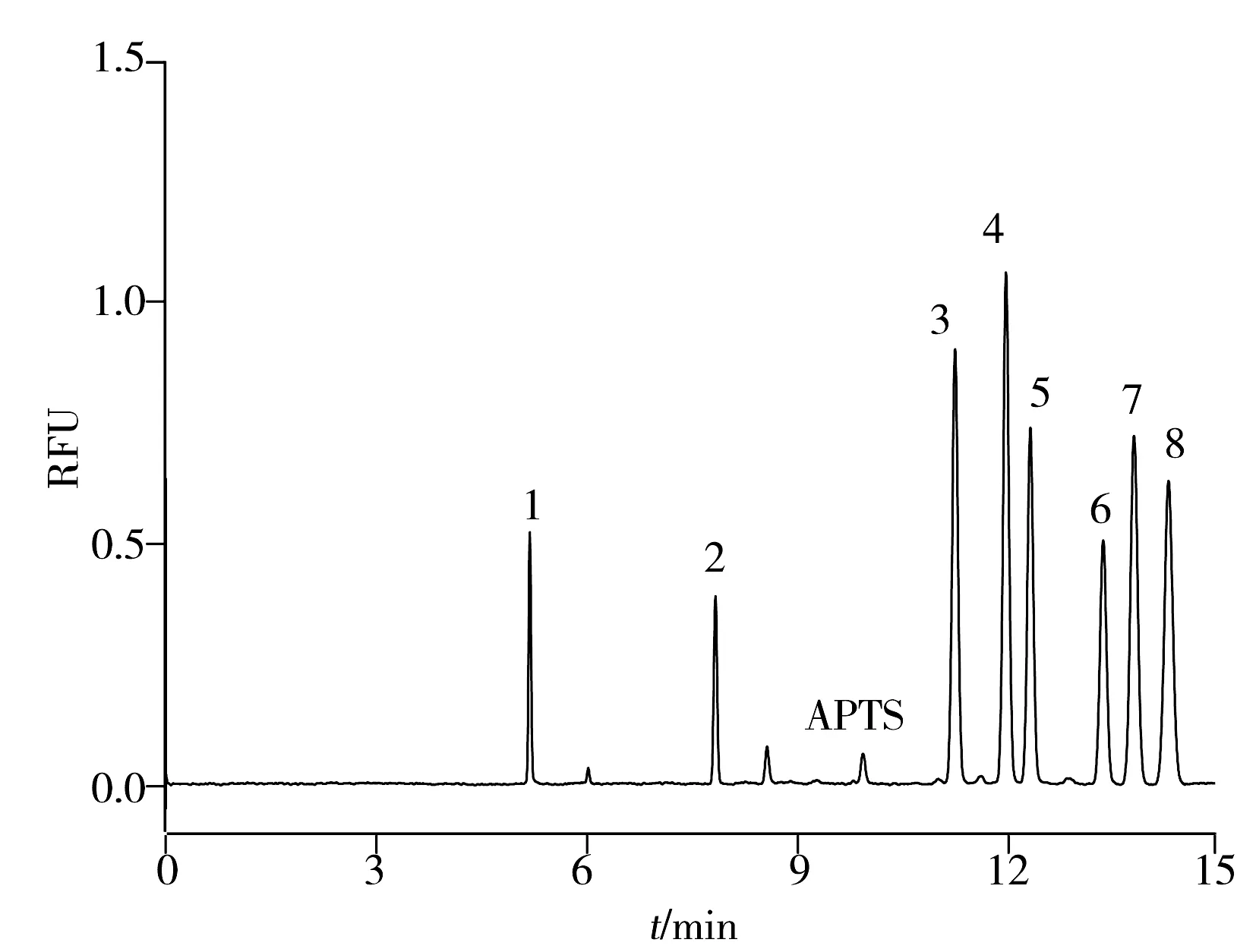
图1 单糖混合标样的毛细管区带电泳图Fig.1 Electropherogram of seven monosaccharide standards1. FS;2.Mal;3.Rha;4.Man;5.Glu;6.Xyl;7.Ara;8.Gal
2.1.2缓冲液及pH值对糖分离的影响考察了磷酸-磷酸二氢钠、Tris-磷酸、醋酸-醋酸钠、硼砂-磷酸以及硼砂-氢氧化钠等不同缓冲体系对待测物分离度和迁移时间的影响。结果表明:由于硼酸盐能与待测物螯合生成配位阴离子来增加溶解度,仅在硼砂缓冲体系中,可减少因吸附造成的峰拖尾等影响,各组分实现了基线分离。在pH 9.2~11.0范围内研究了pH值对被测物分离度的影响,发现pH 10.03时各组分能实现基线分离,故最终选择硼砂-氢氧化钠缓冲体系(pH 10.03)为运行缓冲溶液。
2.1.3方法学的考察在最优的分离条件下,考察了该方法的重复性。以标准连续进样结果计算峰面积的相对标准偏差(RSD),如表1所示,日内和日间的RSD均小于3.0%,表现出良好的重复性。取6种不同浓度的单糖溶液(每种单糖的质量浓度均在0.005 ~0.25 mg/L之间)进行电泳实验,每个样品重复进样4~6次,以各化合物的质量浓度(X,mg/L)与峰面积(Y)绘制标准曲线,再通过回收率验证方法的准确性。结果表明,7种单糖的线性良好,回收率为88.0% ~ 102%,表明该方法的准确性较好。
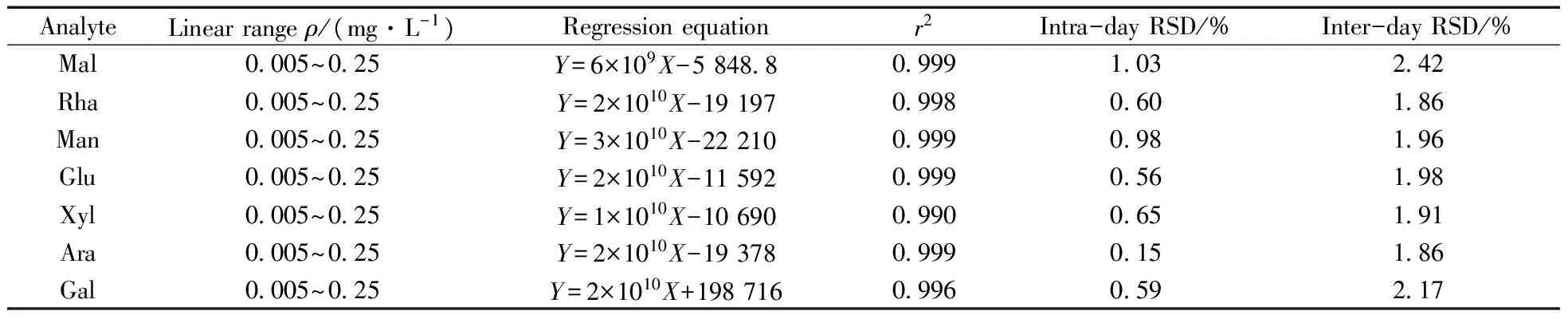
表1 各组分的线性范围、回归方程、相关系数及重复性数据Table 1 Linear range,regression equation,correlation coefficient(r2) and repeatability of the analytes
2.1.4中药多糖特征指纹图谱的建立与分析取药用植物及中药注射液按“1.2”方法处理,经APTS衍生化后进行CZE分析,记录多糖水解产物15 min的毛细管区带电泳图。中药制剂(以批号150607热毒宁注射剂为例)与药用植物多糖(以批号Y Sample 6金银花为例)水解样品中糖分离测定的电泳图谱见图2。通过与单糖标准品进行比较,对实际样品中单糖组分进行了测定。其中,热毒宁注射剂中只检测出葡萄糖和半乳糖(图2A),质量浓度分别为27.58、1.97 mg/mL。金银花中检测出5种单糖(图2B),各单糖的组成比为Man∶Glu∶Xyl∶Ara∶Gal=1.08∶1∶5.14∶2.92∶3.46。对热毒宁注射液不同批次的中药制剂按本方法进行测定发现,其所含葡萄糖的浓度范围为2.03~27.58 mg/mL;半乳糖的最高浓度为1.97 mg/mL,部分热毒宁注射剂不含半乳糖。而对药用植物金银花采用不同提取条件分离纯化后的多糖按本方法进行分析测试发现,不同提取条件对单糖含量和种类影响较大,有一个样品中只含有阿拉伯糖。所以,在生产中对药用植物多糖的日常监控显得十分重要。

2.2 人血清中N-糖链指纹图谱的建立
2.2.1毛细管凝胶电泳(CGE)条件的优化Maltodextrin Ladder是不同聚合度的葡聚糖混合物,聚合度为1~20。APTS标记后常作为复杂糖链电泳分析的外标,不同聚合度的寡糖的迁移时间通过拟合多项式分布曲线进行相关分析。然后将迁移时间转化为葡萄糖单元(Glucose units,GUs),可以作为参考标准校正系统差异,使数据更加可靠。每个糖链的GU值是可重复的,GU 值增量的变化可以预测在原来糖链基础上增加的单糖个数。

考察了磷酸-磷酸二氢钠、乙酸、乙酸铵、Tris-乙酸等不同缓冲体系对寡糖链的分离度和迁移时间的影响,结果发现乙酸铵体系的分离度最好。以不同寡聚度的葡聚糖梯度混合物(Maltodextrin ladder)为标准品进行分离,对盐浓度、添加剂的类型及毛细管柱长进一步优化。结果发现加入聚环氧乙烷(PEO)有利于分离,特别是对较大的寡糖链效果较好。与Mw10000的PEO 相比,Mw20000 PEO的峰形更对称,半峰宽更小。实验得到的最优背景电解质为含有0.4% PEG 20000的25 mmol/L NH4Ac(pH 4.75),分离电压为20 kV,柱温为25 ℃。在上述分离条件下,对葡聚糖的聚合度值(Glucose units,GUs)与各组分电泳迁移时间进行相关分析,通过拟合多项式分布曲线发现其相关性良好,相关系数(r2)为0.99。
2.2.2人血清中N-糖链特征指纹图谱的建立与分析蛋白的糖基化与疾病的发生和发展密切相关。血清作为人体最重要的体液,在临床诊断和疾病的监控中应用广泛。因此,建立人血清中N-糖链的指纹图谱,是进一步实现疾病中N-糖链差异性分析的基础。在优化实验条件下,对人血清中N-糖链经APTS衍生化进行分析(图3)。由于所有的N-糖链均含有1个共同的结构花样(motif),称为核心五糖(Man3GlcNAc2)。因此,N-糖链的迁移时间集中在12~24 min,含量最高组分的峰高为7.50 RFU。以相对荧光强度大于0.2作为参考标准,对相应的峰进行编号,共得到17个组分。
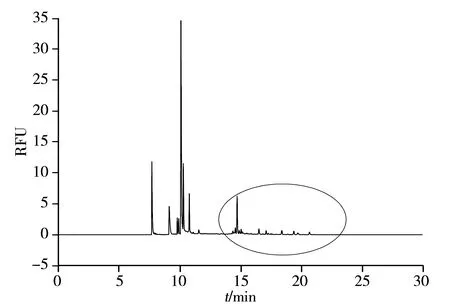
3 结 论
本文基于APTS衍生化/CE-LIF分析,建立了中药中单糖以及人血清中N-糖链的指纹图谱。以硼砂缓冲液为背景电解质,15 min内实现了7种中性单糖标准品的快速分离,并对中药制剂及药用植物多糖中单糖组成、含量进行了分析。该方法不仅为中药注射液中糖类物质含量的监控提供了一种快速、灵敏、有效的分析方法,也对注射液提取工艺的优化具有指导意义。同时本文采用实验室自制的中性PVA涂层柱,建立了人血清中N-糖链的指纹图谱。对μL级的血清样品进行电泳分析,初步分离得到了17个N-糖链组分。这为探索血清蛋白的糖基化变化提供了分析方法,有望用于生物标记物的发现。
[1] Chen X,Zhang Y,Zhang J B.Chin.J.New.Drugs(陈旋,张翼,张剑波.中国新药杂志),2007,16(13):1000-1005.
[2] Hart G W,Copeland R J.Cell,2010,143(5):672-676.
[3] Higel F,Seidl A,Sörgel F,Friess.Eur.J.Pharm.Biopharm.,2016,100:94-100.
[4] Hmiel L K,Brorson K A,Michael I I.Anal.Bioanal.Chem.,2015,407(1):79-94.
[5] Wang C J,Wang Z F.Chin.Bull.LifeSci.(王承健,王仲孚.生命科学),2011,23(6):569-577.
[6] Pu J H,Zhao X,Han W W,Bai M Y.J.Instrum.Anal.(蒲江华,赵峡,韩文伟,白明月.分析测试学报),2017,36(1):145-150.
[7] Guttman A.TrendsinAnal.Chem.,2013,48:132-143.
[8] Szabo Z,Guttman A,Bones J,Karger B L.Anal.Chem.,2011,83:5329-5336.
[9] Lin Y F,He J,Cao L L,Hu X Z,Yu J X.J.Instrum.Anal.(林雁飞,何进,操丽丽,胡小钟,余建新.分析测试学报),2005,24(4):74-76.
[10] Chen N D,Meng Y F,Yao H J,Cao C Y,Chen C.J.Chin.Med.Mater.(陈乃东,孟云飞,姚厚军,曹彩芸,陈辰.中药材),2015,38(8):1607-1610.
[11] Wang W B,Wang L,Wang X,Yu C F,Zhang F,Liu C Y,Chen W,Li M,Gao K.Chin.J.New.Drugs(王文波,王兰,王馨,于传飞,张峰,刘春雨,陈伟,李萌,高凯.中国新药杂志),2015,24(20):2312-2316.
[12] Bai X W,Wang Y H,Chen D M,Tian L.J.Anal.Sci.(白新伟,王毅红,陈定梅,田玲.分析科学学报),2016,32(2):265-268.
[13] Szigeti M,Guttman A.Anal.Chem.,2017,89(4):2201-2204.
[14] Callewaert N,Geysens S,Molemans F,Contreras R.Glycobiology,2001,11(4):275-281.
[15] Vanderschaeghe D,Szekrényes,Wenz C,Gassmann M,Naik N,Bynum M,Yin H F,Delanghe J,Guttman A,Callewaert N.Anal.Chem.,2010,82(17):7408-7415.
Research on Capillary Electrophoresis Fingerprints of Polysaccharides in Traditional Chinese Medicines and N-Glycome in Human Serum Glycoprotein
LI Feng1*,GUAN Yue-qing2,ZHANG Yan-mei3,KANG Jing-wu2
(1.School of Chemical Engineering,Xi’an University,Xi’an 710065,China;2.State Key Laboratory of Bioorganic and Natural Products Chemistry,Shanghai Institute of Organic Chemistry,Chinese Academy of Sciences,Shanghai 200032,China;3.Institute for Agri-food Standards and Testing Technology,Shanghai Academy of Agricultural Sciences,Shanghai 200032,China)
A capillary electrophoresis with laser induced fluorescence detection(CE-LIF) method was developed for the determinations of polysaccharides in traditional Chinese medicines(TCMs) and N-glycome in human serum glycoprotein in this paper.After pre-column derivatized with 9-aminopyrene-1,4,6-trisulfonate(APTS) by reductive amination,the seven monosaccharides were sufficiently separated within 15 min.In the analysis of real samples,the average recoveries for the analytes were all in the range of 88.0%-102% with relative standard deviations(RSDs) lower than 3.0%.The method is sensitive,accurate and reliable,and is suitable for the determination of monosaccharide composition in Chinese medicinal preparation.Meanwhile,fingerprints of N-glycome in human serum were established based on capillary gel electrophoresis(CGE).A poly(vinly alcohol) neutrally coated column was used to suppress the electroosmotic flow which would increase the analysis time,and the excellent separation of glycan isomers was realized.5 μL human sera was analyzed and a total of 17 fractions were isolated(RFU>0.2).The analysis of N-glycan profile has made it possible to research the glycosylation alterations of complex biological fluid.
capillary electrophoresis with laser-induced fluorescence;polysaccharides from traditional Chinese medicines;N-glycosylation;glycoprotein
2017-06-19;
2017-08-31
西安市科技计划项目(2016CXWL18);陕西省教育厅专项科研计划项目(17JK1127)
*
李 凤,博士,讲师,研究方向:毛细管电泳与药物分析,Tel:029-88279060,E-mail:lifeng_sunny@163.com
10.3969/j.issn.1004-4957.2017.12.017
O657.8;R917
A
1004-4957(2017)12-1516-06
