人肝细胞癌细胞DNA甲基化谱的检测及分析
2017-12-19孙宁张佳林张城硕周翔宇陈保民
孙宁,张佳林,张城硕,周翔宇,陈保民
(中国医科大学附属第一医院肝胆外科暨器官移植科,沈阳 110001)
人肝细胞癌细胞DNA甲基化谱的检测及分析
孙宁,张佳林,张城硕,周翔宇,陈保民
(中国医科大学附属第一医院肝胆外科暨器官移植科,沈阳 110001)
目的检测人肝细胞癌细胞的DNA甲基化谱,明确肝细胞癌细胞中差异甲基化位点和基因的表达分布情况,进一步探讨DNA异常甲基化与肝细胞癌发生发展的关系。方法使用DNA甲基化芯片(Infinium Human Methylation 450K BeadChip)检测人肝细胞癌细胞Huh7和人永生化肝细胞L02的甲基化谱,并对检测结果进行生物学分析。结果共检测到差异性甲基化位点102 254个,差异性甲基化基因26 511个,甲基化相关信号通路43个,其中57.3%的高甲基化CpG位点和39.4%的低甲基化CpG位点的甲基化差异程度≥50%,筛选后确定了3 222个显著高甲基化基因和2 204个显著低甲基化基因。结论在Huh7和L02细胞中存在大量差异性甲基化CpG位点及基因,Huh7细胞中可检测到大量抑癌基因DNA的异常高甲基化,提示DNA异常甲基化与肝细胞癌的发生发展密切相关。
肝细胞癌; 甲基化谱; 生物信息学分析
原发性肝癌(primary liver cancer,PLC)是我国常见的消化系统恶性肿瘤,2015年我国PLC新发病例约为46.6万人,发病率位列全部恶性肿瘤第4位,全年死亡病例约为42.2万,致死率位列全部恶性肿瘤第3位[1]。肝细胞癌(hepatocellular carcinoma,HCC)是PLC最常见的类型,导致HCC发生发展的因素有很多,如慢性病毒性肝炎、酒精性肝硬化、黄曲霉素的摄入等,但其具体发病机制仍不明确,因此,研究其发生发展机制对寻找其新的治疗方案至关重要。
随着基因组领域研究的不断深入,表观遗传学改变对疾病的影响逐渐引起研究者的关注。DNA异常甲基化是表观遗传学中最常见也是最重要的修饰,其异常改变被认为是导致包括HCC在内的多种肿瘤发生发展的主要因素[2]。研究[3-4]发现,基因的异常甲基化可影响其转录活性,使其表达下调,尤其是抑癌基因的异常高甲基化常可引起其抑癌功能的失活,最终导致肿瘤的发生。
高通量甲基化芯片Infinium Human Methylation 450K BeadChip能够对人全基因组45万个甲基化位点进行检测,覆盖了上一代27K甲基化芯片90%的位点,并同时增加了CpG岛以外的CpG位点、人类干细胞非CpG甲基化位点、正常组织与肿瘤(多种癌症)组织差异甲基化位点、编码区以外的CpG岛、miRNA启动子区域和已通过GWAS的疾病相关区域的位点。因此,本研究使用Infinium Human Methylation 450K BeadChip对人HCC细胞系Huh7和人永生化肝细胞系L02的DNA甲基化谱进行检测并对其进行分析,筛选出具有显著差异的甲基化CpG位点和基因,明确HCC细胞中差异甲基化CpG位点的表达和分布情况,进一步探讨DNA异常甲基化与HCC发生发展的关系。
1 材料与方法
1.1 材料
人永生化肝细胞L02、人HCC细胞系Huh7均购自中国科学院细胞库;DNA提取试剂盒(QIAamp DNA Micro Kit)购自美国QIAGEN公司;甲基化修饰试剂盒购自沈阳百创特公司;人全基因组甲基化芯片(Infinium Human Methylation 450K BeadChip) 购自美国Illumina公司。
1.2 细胞培养
L02细胞及Huh7细胞分别单层贴壁生长于RPMI-1640培养液或DMEM培养液中,培养液中均含10%胎牛血清、青霉素G (100 U/L)、链霉素(100 μ g/L),于37 ℃、5% CO2恒温密闭式培养箱中培养,隔日换液,取对数生长期细胞进行后续实验。
1.3 DNA提取及甲基化修饰
用0.25%含EDTA胰酶消化细胞,收集细胞沉淀,取20 μ L Proteinase K 加入到1.5 mL Eppendorf管内,按DNA提取试剂盒说明书中的操作步骤提取Huh7、L02细胞DNA,Nano Drop核酸蛋白分析仪检测DNA提取纯度和浓度,若OD260nm/OD280mn在1.7~2.0之间则符合实验要求,否则样品需要重新提取。将提取的Huh7、L02细胞DNA按照甲基化修饰试剂盒说明书中的操作步骤进行甲基化修饰。
1.4 芯片杂交、扫描及数据分析
按照Illumina公司提供的步骤进行芯片杂交;数据扫描使用manifest文件,采用Illumina公司官方提供的Methylation Module of GenomeStudio software using Methylation v1.9软件对芯片数据进行分析。β-value [β = intensity of the methylated allele(M)/(intensity of the unmethylated allele + intensity of the methylated allele + 100)]代表CpG位点的甲基化水平,数值越大,越趋近于1,甲基化程度越高[5];β-difference (绝对值)为表达差异性分数。本研究使用empirical bayes moderated t 检验及Bonferroni校正来识别Huh7细胞和L02细胞之间的差异性甲基化CpG位点。初步筛选差异性甲基化CpG位点的纳入标准为经过校正的P值(Adjust P)≤0.05,同时表达差异性分数(|β-difference|)≥0.2,被定义为具有统计学意义。而Adjust P > 0.05和CpG覆盖<95%则被排除。另外,为进一步筛选具有显著差异的甲基化CpG位点,本研究使用了额外的过滤标准: (1) Adjust P ≤0.05,P ≤1.06×10-7;(2)显著的高甲基化位点,即差异性甲基化水平>20%,L02甲基化水平<25%;(3)显著低甲基化位点,即差异性甲基化水平>20%,Huh7甲基化水平<25%。
2 结果
2.1 Huh7细胞全基因组差异性甲基化位点表达情况
本研究中,共检测到差异性甲基化位点102 254个,差异性甲基化基因26 511个。其中,高甲基化CpG位点62 702个(61.3%),包含12 665个高甲基化基因;低甲基化位点39 552个(38.7%),包含13 846个低甲基化基因。另外,在CpG岛中,检测到了差异性CpG位点41 178个(40.3%);在CpG shores检测到了差异性甲基化CpG位点21 150个(20.7%);在CpG shelves检测到9 030个(8.8%)差异性甲基化CpG位点。见表1,图1。
2.2 差异性甲基化CpG位点甲基化差异程度的分布
本研究结果显示,有35 937个(57.3%)高甲基化CpG位点和15 587个(39.4%)低甲基化CpG位点的甲基化差异程度≥50%;18 529个(29.5%)高甲基化CpG位点和14 177个(35.8%)低甲基化CpG位点的甲基化差异程度≥30%但<50%;8 236个(13.1%)高甲基化CpG位点和9 788个(24.8%)低甲基化CpG位点的甲基化差异程度<30%但≥20%。见表2。

表1 总体差异性甲基化CpG位点分布 [n(%)]Tab.1 Distribution of all the differentially methylated CpG sites [n(%)]
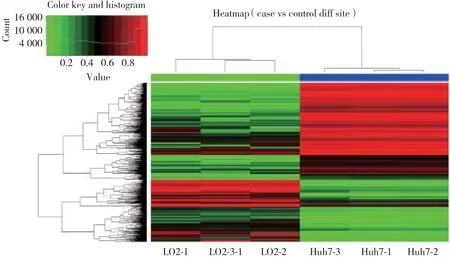
图1 差异性甲基化CpG位点的聚类分析Fig.1 Hierarchical cluster analysis of all differentially methylated CpG sites between Huh7 cells and L02 cells
2.3 显著差异性甲基化CpG位点分布及差异性甲基化基因筛选
通过使用严格的筛选条件对差异性甲基化CpG位点进行筛选后,确定了5 285个明显的高甲基化CpG位点(包括3 222个基因)和2 659个明显的低甲基化CpG位点(包括2 204个基因)。见表3~5,图2。
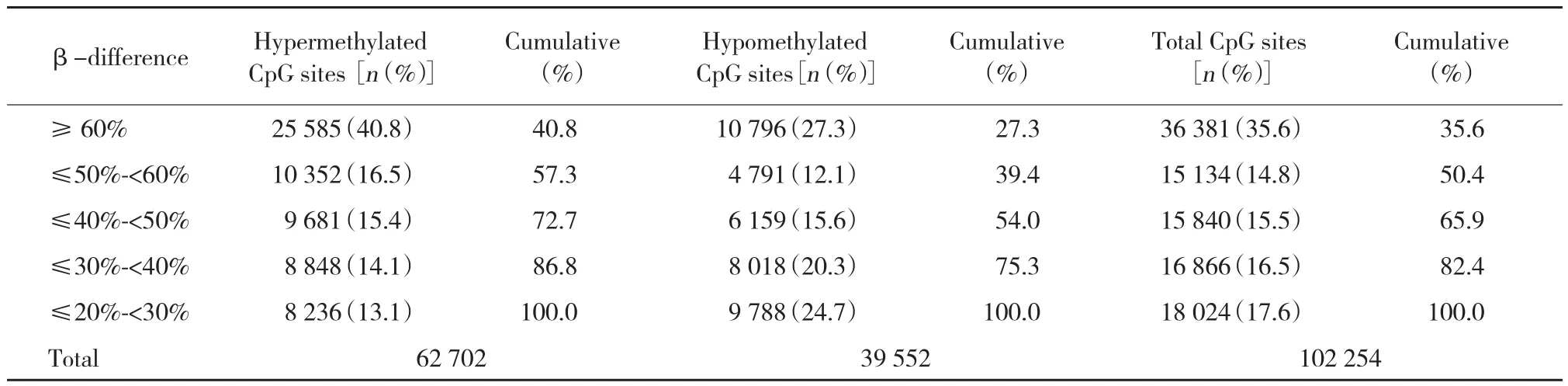
表2 差异性甲基化CpG位点甲基化差异程度的分布Tab.2 Distribution of all the differentially methylated CpG sites between Huh7 and L02 cells based on methylation status
2.4 差异性甲基化基因的GO富集分析和KEGG通路分析
本研究使用GO富集分析(http://www.geneontol ogy.org/)和 KEEG 通路分析 (the Kyoto Encyclopedia of Genes and Genomes,http://www.genome.jp/kegg/)对差异性位点进行相关通路分析。GO富集的结果如下:2 107个差异甲基化基因富集于9个生化过程相关功能集,富集最明显的包括负调控的细胞增殖,子宫内的胚胎发育等;13 351个差异甲基化基因富集于17个分子功能相关功能集,富集最明显的包括蛋白结合,金属离子结合等;18 041个差异甲基化基因富集于与细胞成分有关的功能集,富集最明显的主要是细胞核、细胞质等。KEGG通路分析显示43个信号通路涉及5 195个差异甲基化的基因,这些基因主要富集于代谢通路、癌症相关通路、MAPK信号通路、Wnt 信号通路、VEGF 信号通路和 p53 信号通路等,见表6。
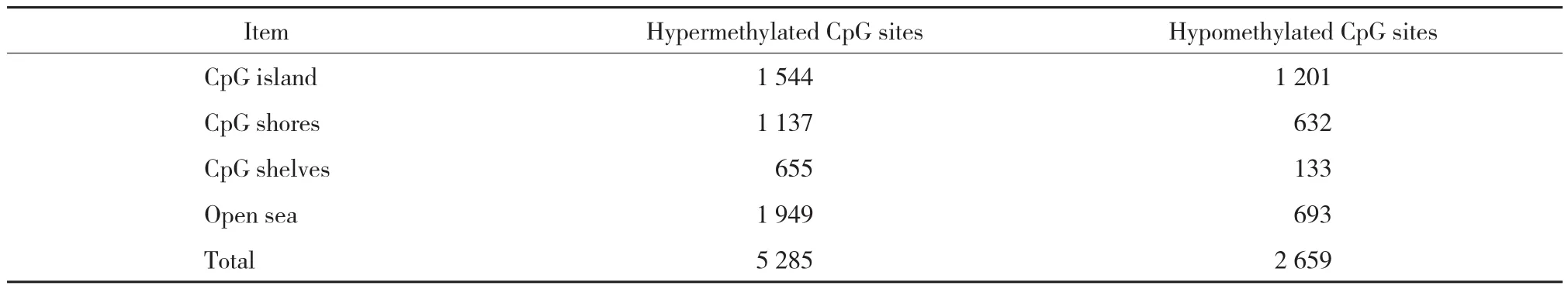
表3 显著差异性甲基化位点分布情况(n)Tab.3 Distribution of significantly differentially methylated CpG sites in Huh7 cells when compared with L02 cells(n)
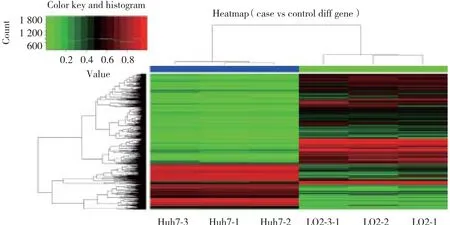
图2 差异性甲基化CpG基因的聚类分析Fig.2 Hierarchical cluster analysis of all differentially methylated genes between Huh7 cells and L02 cells

表4 前10个显著差异高甲基化基因Tab.4 Top 10 significant hypermenthylated CpG sites and genes within DMRs in Huh7 cells compared with L02 cells
3 讨论
近年来,基因的DNA异常甲基化被认为是导致多种肿瘤发生的重要因素[6-7]。既往针对HCC相关基因的DNA甲基化研究虽然能够为HCC的诊断和预后评估提供一些新的甲基化标记物,但关于HCC DNA甲基化谱的研究甚少。因此,本研究利用Infinum Human Methylation 450K芯片技术对HCC细胞的DNA甲基化谱进行检测,并对结果进行分析。

表5 前10个显著差异低甲基化基因Tab.5 Top 10 significant CpG hypomethylated sites and genes within DMRs in Huh7 cells compared with L02 cells
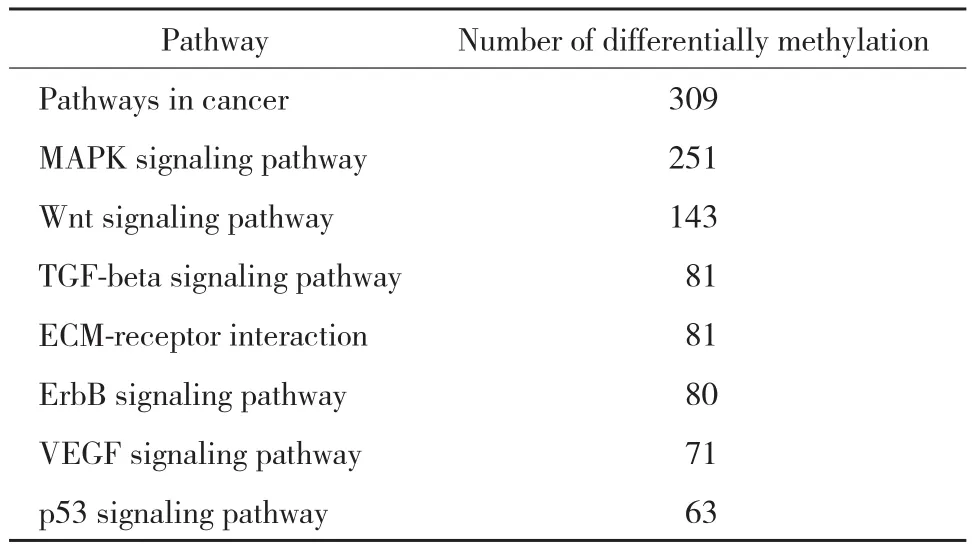
表6 主要HCC相关通路中差异甲基化基因数目Tab.6 Number of differentially methylated gene in HCC related pathway
Infinium Human Methylation 450K甲基化芯片涵盖99%在注释的RefSeq基因启动子区的多个位点、5’-UTR、第一外显子、3’-UTR等重要区域[8]。在本研究结果中,共检测到102 254个差异性甲基化CpG位点,并且高甲基化CpG位点(62 702,61.3%)明显多于低甲基化CpG位点(39 552,38.7%),表明在Huh7细胞中DNA异常甲基化是一种常见事件,而且发生频率较高。同时,还发现显著高甲基化CpG位点在CpG岛的分布要多于显著低甲基化位点(1 544 vs.1 201),这与之前其他相关的DNA甲基化基因分析研究[9-12]结果一致。
有研究[13-17]认为,DNA异常甲基化不仅发生在CpG岛,也可能发生在CpG shores和CpG shelves等区域,并同样能够导致抑癌基因的功能失活。本研究还发现在CpG shores的确分布有一定数量的显著差异CpG位点,且高甲基化CpG位点多于低甲基化位点(1 137 vs 632),而在CpG shelves,高甲基化CpG位点更明显多于低甲基化CpG位点(655 vs 133)。本研究结果不仅进一步证实了其他研究的结论,同时也提示,在HCC细胞中能够检测到大量DNA异常高甲基化位点,这些异常位点很可能与HCC相关抑癌基因功能失活有关,参与了HCC的发生发展。
另外,本研究通过对差异性甲基化基因的GO分析和KEGG通路分析发现DNA的异常甲基化能够影响细胞周期相关蛋白p16(Ink4a)、p53、TGF-β/SMAD信号等经典癌症相关通路[18]。在本研究中,虽然对HCC细胞DNA的甲基化谱进行了检测及分析,但是仍需要进一步的实验研究对现有的芯片结果进行验证,继续深入探讨DNA异常甲基化在HCC发生发展中所起的作用及其临床意义。现有研究[19-20]已证实,在HCC中,Wnt通路的激活与抑癌基因DNA异常高甲基化有关,Erb受体信号途径和MAPK信号通路也已被证实其相关蛋白质经过表观遗传学异常修饰后活性改变,从而导致了肿瘤生长和转移[16,21],这些研究对下一步芯片结果的验证均具有一定的指导意义。
综上所述,本研究应用高通量甲基化芯片测序技术对Huh7细胞和L02细胞的全基因组DNA甲基化模式进行了探讨,初步明确了差异性甲基化CpG位点/区域/基因在HCC细胞中的表达频率和分布情况,为今后HCC相关的甲基化研究提供了更多信息。本研究结果中的显著差异位点和基因仍需进行验证,其相关分子机制也有待进一步研究,以继续深入探究DNA甲基化与HCC发生发展的关系,也为今后HCC的去甲基化治疗奠定基础。
[1] CHEN W,ZHENG R,BAADE PD,et al. Cancer statistics in China,2015[ J]. CA Cancer J Clin,2016,66(2):115-132. DOI:10.3322/caac.21338.
[2] KULIS M,ESTELLER M. DNA methylation and cancer[ J]. Adv Genet,2010,70:27-56. DOI:10.1016/B978-0-12-380866-0.60002-2.
[3] HERMAN JG,BAYLIN SB. Gene silencing in cancer in association with promoter hypermethylation[ J]. N Engl J Med,2003,349( 21):2042-2054. DOI:10.1056/NEJMra023075.
[4] ESTELLER M. Epigenetic gene silencing in cancer:the DNA hypermethylome[ J]. Hum Mol Genet,2007,16( 1):R50-R59. DOI:10.1093/hmg/ddm018.
[5] BIBIKOVA M,LE J,BARNES B,et al. Genome-wide DNA methylation profiling using Infinium(R) assay[ J]. Epigenomics,2009,1( 1):177-200. DOI:10.2217/epi.09.14.
[6] JONES P A,TAKAI D. The role of DNA methylation in mammalian epigenetics[ J]. Science,2001,293( 5532):1068-1070. DOI:10.1126/science.1063852.
[7] ESTELLER M. Epigenetics in cancer[ J]. N Engl J Med,2008,358(11):1148-1159. DOI:10.1056/NEJMra072067.
[8] BIBIKOVA M,BARNES B,TSAN C,et al. High density DNA methylation array with single CpG site resolution[ J]. Genomics,2011,98(4):288-295. DOI:10.1016/j.ygeno.2011.07.007.
[9] GAO W,KONDO Y,SHEN L,et al. Variable DNA methylation patterns associated with progression of disease in hepatocellular carcinomas[ J]. Carcinogenesis,2008,29( 10):1901-1910. DOI:10.1093/carcin/bgn170.
[10] SHIN SH,KIM BH,JANG JJ,et al. Identification of novel methylation markers in hepatocellular carcinoma using a methylation array[J]. J Korean Med Sci,2010,25( 8):1152-1159. DOI:10.3346/jkms.2010.25.8.1152.
[11] AMMERPOHL O,PRASTCHKE J,SCHAFMAYER C,et al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma[ J]. Int J Cancer,2012,130( 6):1319-1328. DOI:10.1002/ijc.26136.
[12] KOHLES N,NAGEL D,JUNGST D,et al. Prognostic relevance of oncological serum biomarkers in liver cancer patients undergoing transarterial chemoembolization therapy[ J]. Tumour Biol,2012,33(1):33-40. DOI:10.1007/s13277-011-0237-7.
[13] YATES DR,REHMAN I,MEUTH M,et al. Methylational urinalysis:a prospective study of bladder cancer patients and age stratified benign controls[ J]. Oncogene,2006,25( 13):1984-1988. DOI:10.1038/sj.onc.1209209.
[14] DUZIEC E,MIAH S,CHOUDHRY HM,et al. Hypermethylation of CpG islands and shores around specific microRNAs and mirtrons is associated with the phenotype and presence of bladder cancer,clinical cancer research[ J]. Clin Cancer Res,2011,17( 6):1287-1296.DOI:10.1158/1078-0432.CCR-10-2017.
[15] DOI A,PARK IH,WEN B,et al. Differential methylation of tissueand cancer-specific CpG island shores distinguishes human induced pluripotent stem cells,embryonic stem cells and fibroblasts[ J]. Nat Genet,2009,41( 12):1350-1353. DOI:10.1038/ng.471.
[16] IRIZARRY RA,LADD-ACOSTA C,WEN B,et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores[ J]. Nat Genet,2009,41(2):178-186. DOI:10.1038/ng.298.
[17] OGOSHI K,HASHIMOTO S,NAKATANI Y,et al. Genome-wide profiling of DNA methylation in human cancer cells[ J]. Genomics,2011,98( 4):280-287. DOI:10.1016/j.ygeno.2011.07.003.
[18] KUO KK,JIAN SF,LI YJ,et al. Epigenetic inactivation of transforming growth factor-beta1 target gene HEYL,a novel tumor suppressor,is involved in the P53-induced apoptotic pathway in hepatocellular carcinoma[ J]. Hepatol Res,2015,45( 7):782-793. DOI:10.1111/hepr.12414.
[19] UMER M,QURESHI SA,HASHMI ZY,et al. Promoter hypermethylation of Wnt pathway inhibitors in hepatitis C virus-induced multistep hepatocarcinogenesis[ J]. Virol J,2014,11:117. DOI:10.1186/1743-422X-11-117.
[20] DING SL,YANG ZW,WANG J,et al. Integrative analysis of aberrant Wnt signaling in hepatitis B virus-related hepatocellular carcinoma[ J]. World J Gastroenterol,2015,21( 20):6317-6328. DOI:10.3748/wjg.v21.i20.6317.
[21] STEFANSKA B,CHEISHVILI D,SUDERMAN M,et al. Genome-wide study of hypomethylated and induced genes in patients with liver cancer unravels novel anticancer targets[ J]. Clin Cancer Res,2014,20( 12):3118-3132. DOI:10.1158/1078-0432.CCR-13-0283.
Exploring Genome-wide Profiles of DNA Methylation in Human Hepatocellular Carcinoma Cells via Bioinformatics Analysis
SUN Ning,ZHANG Jialin,ZHANG Chengshuo,ZHOU Xiangyu,CHEN Baomin
(Department of Hepatobiliary and Transplantation Surgery,The First Hospital,China Medical University,Shenyang 110001,China)
ObjectiveTo detect the genome-wide profiles of DNA methylation in human hepatocellular carcinoma (HCC) cells and to identify the distribution of differentially methylated sites and genes in order to explore the relationship between aberrant DNA methylation and hepatocellular carcinoma.MethodsThe Infinium Human Methylation 450K BeadChip was used to identify the genome-wide aberrant DNA methylation profiles in Huh7 and L02 cell lines.ResultsTotally 102 254 differentially methylated CpG sites and 26 511 genes,involving 43 signaling pathways,were detected when Huh7 and L02 cell lines were compared. The absolute β-difference in 57.3%of the hypermethylated CpG sites and 39.4% of the hypomethylated CpG sites was reported to be ≥ 50%. A total of 3 222 hypermethylated genes and 2 204 hypomethylated genes were identified.ConclusionWe detected many aberrant methylated sites and genes in HCC cells. The abnormal DNA methylation exhibits an important role in the occurrence and development of HCC.
hepatocellular carcinoma; methylation profiles; bioinformatics analysis
R735.7
A
0258-4646(2017)12-1111-06
http://kns.cnki.net/kcms/detail/21.1227.R.20171130.1810.022.html
10.12007/j.issn.0258-4646.2017.12.012
沈阳市科学技术计划(F13-F13-212-9-00)
孙宁(1986-),女,医师,博士.
张佳林,E-mail:jlzcmu@126.com
2017-03-28
网络出版时间:2017-11-30 18:10
(编辑 王又冬)
