蛋白酶激活受体2/P38MAPK信号通路在缺血后处理对抗老年糖尿病大鼠心肌缺血再灌注损伤中的作用*
2017-12-15张秀娥戚本玲
张秀娥 成 蓓 何 平 戚本玲
蛋白酶激活受体2/P38MAPK信号通路在缺血后处理对抗老年糖尿病大鼠心肌缺血再灌注损伤中的作用*
张秀娥 成 蓓#何 平 戚本玲
目的研究蛋白酶激活受体2/P38MAPK(PAR-2/P38MAPK)信号通路在缺血后处理(IPost)对抗老年糖尿病大鼠心肌缺血再灌注(I/R)损伤中的作用。方法22-24月龄雄性Wistar大鼠32只,随机选取24只采用链脲佐菌素(STZ)腹腔注射制备老年糖尿病模型。成模大鼠随机分成3组(每组8只):I/R组、IPost组、PAR-2抑制剂组(PAR-2组),PAR-2组大鼠于缺血处理前1h尾静脉注射PAR-2抑制剂FSLLRY-NH2,其余处理同IPost组;未造模的8只大鼠作为对照组(SC组)。采用黄嘌呤氧化酶法和硫代巴比妥比色法分别测定血清超氧化物歧化酶(SOD)活性及丙二醛(MDA)浓度,TUNEL法检测心肌细胞凋亡率,Western Blotting测定PAR-2及P38MAPK、P-P38MAPK蛋白表达水平。结果I/R组MDA浓度、心肌细胞凋亡率及P-P38MAPK水平均较SC组显著增加(P<0.05或P<0.01),SOD活性、PAR-2蛋白表达水平明显降低(P<0.01)。IPost组SOD活性及PAR-2水平较I/R组显著增加(P<0.05),P-P38MAPK水平、MDA浓度及心肌细胞凋亡率均下降(P<0.01)。PAR-2抑制剂则可抑制IPost作用,使P-P38MAPK蛋白表达水平上调、心肌细胞凋亡率增加、MDA浓度升高、SOD活性降低(P<0.05或P<0.01)。结论IPost对老年糖尿病大鼠心肌I/R损伤有保护作用,其机制与PAR-2/P38MAPK信号通路的调节有关。
蛋白酶激活受体2/P38MAPK;缺血后处理;缺血再灌注损伤;老年糖尿病大鼠
缺血后处理(Ischemic Postconditioning, IPost)是近年发现的一种机体内源性缺血再灌注(Ischemia/Reperfusion, I/R)损伤的保护策略[1],其机制不明。鉴于老年人伴发糖尿病冠心病的发病率逐年升高,严重危害患者健康和生命,故研究IPost对老年糖尿病心肌I/R的保护作用有重要临床价值。我们课题组的前期研究业已表明,IPost对老年糖尿病大鼠心肌I/R确有保护作用,同时发现蛋白酶激活受体2(Protease-Activated Receptor 2,PAR-2)活化可进一步增强此作用[2]。但该效应的具体信号机制并不完全清楚。
P38MAPK信号通路为MAPK家族重要成员,有研究表明,P38MAPK可参与急性心肌梗死后心肌I/R过程,加剧炎症反应,造成心肌损伤[3,4]。但其在IPost拮抗心肌I/R损伤的作用及与PAR-2活化的关系尚未见文献报道。本研究通过建立老年糖尿病大鼠I/R模型,探讨PAR-2如何调节P38MAPK信号通路,从而实现IPost对抗老年糖尿病大鼠心肌I/R损伤的作用。
1 材料与方法
1.1 主要试剂与仪器
兔抗P38MAPK抗体、兔抗P-P38MAPK抗体、兔抗PAR-2抗体(美国Santa Cruz公司,批号sc-81621、 sc-166182、sc-400333), 兔抗β-Tubulin抗体(武汉三鹰生物技术有限公司,批号20150718),山羊抗兔二抗(北京中杉生物技术公司,批号20140630),链脲佐菌素(STZ, 美国Sigma公司,批号S0084),PAR-2抑制剂FSLLRY-NH2粉剂(上海生工生物工程技术服务有限公司,批号E5971860002); TUNEL检测试剂盒(武汉博士德生物制品有限公司,批号20140306);血清超氧化物歧化酶(SOD)和丙二醛(MDA)检测试剂盒(南京建成生物工程公司,批号20140115,20140117)。Quantity One 图像分析系统(美国BioRad公司,型号170-8170),H-X小型动物呼吸机(城都泰盟科技有限公司,型号HX-300S),心电监护仪(上海玉研科学仪器有限公司), CIAS -1000细胞图像分析仪(北京大恒图像视觉有限公司)。
1.2 实验动物及复制糖尿病模型
健康清洁级雄性Wistar大鼠32只,22-24月龄,体重350-550g,由华中科技大学同济医学院实验动物学部提供。适应性饲养5天后,禁食12h,随机取24只,用枸橼酸-枸橼酸钠缓冲液配制的1% STZ溶液一次性腹腔注射(60mg/kg), 72h后禁食4h,断尾取血测血糖,连续3天。24只大鼠血糖浓度均≥16.7mmol/L, 且出现多饮、多食、多尿, 提示糖尿病大鼠模型诱导成功[5]。成模大鼠适应性饲养1周后, 进行下述分组处理。
1.3 实验动物分组处理
将糖尿病模型大鼠随机分为I/R组、IPost组和PAR-2抑制剂组(PAR-2组),各组均8只,未行造模的8只大鼠平行注射等量生理盐水,作为对照组(SC组)。各组大鼠以1%戊巴比妥钠(40mg/kg)腹腔注射麻醉,仰卧位固定,颈正中切口行气管插管,连接呼吸机辅助呼吸,连接心电监护仪检测心电变化,开胸,暴露心脏。进行如下处理:(1) SC组:于左冠状动脉前降支分支下约2mm处穿一结扎线,不结扎,旷置150min;(2)I/R组:在左冠状动脉前降支分支下约2mm处穿一结扎线,稳定10min后,结扎左前降支30min造成心肌缺血,心电图出现ST段升高时,放松结扎线恢复血流,再灌注120min,至ST段下降50%以上;(3)IPost组:同上结扎左前降支,先缺血30min后,再灌注10s,缺血10s,连续3个循环后, 再灌注120min,至ST段下降50%以上;(4)PAR-2组:所用PAR-2抑制剂FSLLRY-NH2先用双蒸水溶解,配制成1mg/ml浓度溶液,缺血处理前1h尾静脉一次性注射(1ml/kg),之后处理同IPost组;其它三组平行注射与本组等量生理盐水。
1.4 检测指标和方法
1.4.1SOD活性及MDA浓度测定:各组大鼠于末次再灌注后打开腹腔,从下腔静脉抽取静脉血4ml,以4 500r/min离心分离血清,-20℃保存批检。临检时室温复融,取血清样品10μl,黄嘌呤氧化酶法测定SOD活性, 按试剂盒说明书完成相关步骤后,于550nm波长处测定吸光度,计算总SOD活性。另取血清样品0.1ml,硫代巴比妥比色法测定MDA浓度,根据试剂盒说明书完成相关步骤后,于532nm处测定吸光度,计算MDA浓度。
1.4.2TUNEL法检测心肌细胞凋亡:各组大鼠取血后立即处死,取出心脏,沿冠状沟分离左心室心肌,甲醛固定后制备石蜡切片,用TUNEL法检测心肌细胞凋亡,具体操作依照TUNEL细胞凋亡检测试剂盒。镜下观察细胞核中有棕黄色颗粒者为凋亡细胞。采用CIAS-1000细胞图像分析系统,每张切片随机选取5个视野,计算阳性细胞占细胞总数的百分比作为凋亡率。
1.4.3Western Blotting检测心肌细胞PAR-2/P38MAPK/P-P38MAPK蛋白表达:分离部分左心室,经液氮研磨后,取约100mg加入0.8ml冷裂解液,冰浴下超声充分破碎细胞,12 000r/min离心5min,提取总蛋白,Bradford法测定蛋白浓度。取 60μg总蛋白,加等量缓冲液,行SDS-PAGE电泳,再将电泳分离后蛋白电转至PDVF膜上,封闭1h,洗脱后分别加入P38MAPK(1∶10 000)、P-P38MAPK(1∶1 000)、PAR-2(1∶10 000)及β-Tubulin(1∶1 000)一抗孵育过夜,加入山羊抗兔二抗,室温孵育2h,加入ECL,然后对PDVF膜进行显影、定影、灰度扫描,用Quantity One 图像分析系统分析各目标条带与β-Tubulin条带的光密度(OD)比值,用以表示心肌细胞蛋白表达水平(两者呈正比)。
1.5 统计学处理
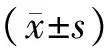
2 结果
2.1 各组血清SOD活性和MDA浓度比较
方差分析显示,各组SOD活性和MDA浓度差异具有统计学意义(均P<0.01)。与SC组比较,I/R组SOD活性显著下降(t=8.756,P<0.01),MDA浓度显著升高(t=-10.578,P<0.01);与I/R组相比,IPost组SOD活性上升(t=-4.565,P<0.01),MDA浓度下降(t=2.453,P<0.05);PAR-2组较IPost组SOD活性下降(t=9.842,P<0.01),MDA浓度升高(t=-11.258,P<0.01)。见表1。

表1 各组检测指标比较均=8)
注:与SC组比较,1)P<0.01;与I/R组比较,2)P<0.05,3)P<0.01;与IPost组相比,4)P<0.01
2.2 各组心肌细胞凋亡率比较
图1显示,SC组未见棕黄色的凋亡心肌细胞,I/R组可见较多凋亡细胞, IPost组凋亡细胞减少,但PAR-2抑制剂可抑制IPost效应,使凋亡细胞增加。定量分析结果为各组心肌细胞凋亡率差异有统计学意义(P<0.01),与SC组比较,I/R组凋亡率明显升高(t=-10.258,P<0.01);与I/R组比较,IPost组凋亡率下降(t=3.105,P<0.01);PAR-2组细胞凋亡率又较IPost组明显升高(t=-5.624,P<0.01)。见表1。
2.3 各组心肌PAR-2 及P-P38MAPK蛋白表达
方差分析显示,各组PAR-2 及P-P38MAPK蛋白OD比值差异均有统计学意义(P<0.01),而各组P38MAPK蛋白OD比值差异无统计学意义(P>0.05)。 与SC组比较,I/R组心肌PAR-2 蛋白OD比值下降(t=3.245,P<0.01),P-P38MAPK 蛋白OD比值升高(t=-5.254,P<0.01);与I/R组比较,IPost组心肌PAR-2蛋白OD比值显著升高(t=-8.542,P<0.01),而心肌P-P38MAPK 蛋白OD比值明显降低(t=4.354,P<0.01);与IPost组比较,PAR-2组PAR-蛋白OD比值降低(t=6.576,P<0.01),P-P38MAPK蛋白OD比值明显升高(t=-5.254,P<0.01)。见图2、表2。

图2 各组PAR-2和P-P38MAPK蛋白电泳结果(Western Blotting)

组 别PAR⁃2P⁃P38MAPKP38MAPKSC组0.96±0.010.98±0.021.01±0.02I/R组0.68±0.011)1.38±0.061)0.98±0.01IPost组1.20±0.032)0.45±0.012)1.02±0.02PAR⁃2组1.01±0.023)0.85±0.043)0.99±0.02F值1.6921.5816.24
注:与SC组比较,1)P<0.01;与I/R组比较,2)P<0.01;与IPost组相比,3)P<0.01
3 讨论
心肌I/R可导致心肌细胞超微结构、功能、代谢及电生理等损害[6]。减轻和消除I/R危害已成为心血管疾病研究的重大课题。较多研究[7,8]认同IPost对心肌I/R损伤具有保护作用,但对其干预机制,尤其针对老年糖尿病干预机制研究涉及较少。本课题组对此进行了有益的探讨。
老年人是冠心病的主要发病人群,特别是糖尿病冠心病患者,其衰老心肌对I/R损伤敏感性更高,梗死面积更大,细胞凋亡更多,也更需要心肌保护。但目前较少有IPost对老年糖尿病心肌I/R损伤保护作用的研究报道。本课题组前期研究[2]发现,IPost 能通过减少心肌细胞凋亡、提高细胞抗氧化力对老年糖尿病大鼠的I/R损伤发挥保护作用,并观察了PAR-2在其中的功效,即给予PAR-2激动剂SLIGRL-NH2于缺血处理前干预,活化PAR-2,可增强IPost拮抗I/R损伤。初步提示,PAR-2活化可能是IPost保护作用的机制之一,但其具体信号通路未及探讨。
P38MAPK是MAPK家族重要成员之一,可调控细胞增殖、分化、凋亡等。研究表明,P38MAPK信号通路的激活在心肌I/R的病理生理进程中具有重要作用[9,10]。PAR-2是否通过P38MAPK信号通路参与IPost对老年糖尿病大鼠I/R损伤的保护作用未见相关报道。本次实验进一步探讨了这一功能的信号通路,即在缺血处理前给予PAR-2抑制剂FSLLRY-NH2干预,结果显示PAR-2抑制剂可降低IPost拮抗I/R损伤作用。PAR-2是IPost抗I/R损伤的调节机制。更为重要的是,本次实验检测了IPost各组大鼠血清P38MAPK蛋白表达水平,并证实,IPost前静脉给予PAR-2抑制剂,使PAR-2蛋白表达下降的同时增加P38MAPK磷酸化(与IPost组相比,P-P38MAP蛋白水平明显升高)继而促进心肌凋亡,降低心肌抗氧化力。本次实验结合上次实验较完整地说明了IPost 中,PAR-2通过P38MAPK磷酸化信号通路实现对老年糖尿病大鼠I/R损伤的保护作用。
综上所述,IPost对老年糖尿病大鼠心肌I/R损伤有保护作用,其机制与PAR-2/P38MAPK信号通路的调节有关,为临床上IPost减轻心肌I/R损伤提供了理论依据,具有重要临床意义。
◀
本文第一作者简介:
张秀娥(1974-),女,汉族,博士,主治医师,研究方向为冠心病的病理生理机制
1 Buchholz B, D Annunzio V,Giani J F, et al. Ischemic postconditioning reduces infarct size through the α1-adrenergic receptor pathway[J]. J Cardiovasc Pharmacol, 2014, 63(6):504-511.
2 张秀娥 ,成 蓓,戚本玲,等. 蛋白酶激活受体2活化与缺血后处理对老年糖尿病大鼠心肌缺血再灌注损伤的保护作用[J]. 微循环学杂志,2017,27(1):1-5.
3 Chen Y, Ba L, Huang W, et al. Role of carvacrol in cardioprotection against myocardial ischemia/reperfusion injury in rats through activation of MAPK/ERK and Akt/eNOS signaling pathways[J]. Eur J Pharmacol, 2017,796(2):90-100.
4 Gao F, Yue TL, Shi DW, et al.p38MAPK inhibition reduces myocardial reperfusion injury via inhibition of endothelial adhesion molecule expression and blockade of PMN accumulation[J]. Cardiovasc Res,2002,53(2):414-422.
5 Charan K, Goyal A, Gupta JK,et al.Role of atrial natriuretic peptide in ischemic preconditioning-induced cardioprotection in the diabetic rat heart[J]. J Surg Res,2016,201(2):272-278.
6 Nagaoka K, Matoba T, Mao Y, et al.A new therapeutic modality for acute myocardial infarction: nanoparticle-mediated delivery of pitavastatin induces cardioprotection from ischemia-reperfusioninjury via activated of PI3K/Akt pathway and anti-inflammation in a rat model [J]. PLoS One, 2015, 10(7): e0132451.
7 Wang ZF, Wang NP, Harmouche S, et al. Postconditioning attenuates coronary perivascular and interstitial fibrosis through modulating angiotensin II receptors and angiotensin-converting enzyme 2 after myocardial infarction[J]. J Surg Res, 2017,211(5):178-190.
8 McGuire JJ,Van Vliet BN, Gimenez J, et al.Persistence of PAR-2vasodilation despite endothelial dysfunction in BPH/2 hypertensive mice[J]. Pflugers Arch, 2007,454(4):535-543.
9 Ran K, Gong ZX,Yang DL,et al. Effect of morphine preconditioning in the delayed phase on the expression of p38Mitogen-activated protein kinase in a rabbit model of myocardial ischemia-reperfusion injury[J]. Genet Mol Res,2015,14(2):6 642-6 648.
10 Wang J,Yang H,Hu X,et al.Dobutamine-mediated hemeoxygenase-1 induction via PI3K and p38MAPK inhibits high mobility group box 1 protein release and attenuates rat myocardial ischemia/reperfusion injury in vivo[J]. J Surg Res,2013,183(2):509-516.
RoleofPAR-2/P38MAPKPathwayinProtectiveEffectsofIschemicPostconditioningagainstMyocardialIschemiaReperfusionInjuryofAgedDiabeticRats
ZHANG Xiu-e, CHENG Bei#, HE Ping, QI Ben-ling
Department of Geriatrics, Institute of Geriatrics,Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022,China;#Corresponding author
Objective: To investigate the role of protease-activated receptor 2(PAR-2)/P38MAPK pathway in protective effects of ischemic postconditioning (IPost) against myocardial ischemia reperfusion(I/R) injury of aged diabetic rats. Method:32 healthy aged(22-24 months) male Wistar rats were obtained. 24 models of aged diabetic rats were induced by using streptozotocin(STZ) and then the diabetic rats were randomly divided into 3 groups (8 animals in each group): ischemia/reperfusion group(I/R group), ischemic postconditioning group(IPost group ) and protease-activated receptor 2 inhibition group(PAR-2 group). FSLLRY-NH2 was injected to inhibit PAR-2 an hour of ischemic treatment ago in PAR-2 group. The remaining treatment was the same as IPost group. In addition, the remaining 8 rats served as saline control group(SC group). The activity of superoxide dismutase(SOD) was detected by xanthine oxidase method and the content of malonaldehyde(MDA) was measured by thiobarbituric acidcolorimetry in each group. Myocardial apoptosis was detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling(TUNEL)staining. The protein levels of PAR-2, P38MAPK and P-P38MAPK were detected by Western blotting.ResultsCompared with SC group, MDA contents, myocardial apoptosis rate and P-P38MAPK level increased significantly in I/R group(P<0.01),the activity of SOD and protein level of PAR-2 decreased significantly (P<0.01). Compared with I/R group, the activity of SOD and protein level of PAR-2 increased in IPost group(P<0.05),the protein level of P-P38MAPK, MDA contents and myocardial apoptosis rate decreased in IPost group(P<0.01). However, the protective effect of IPost was inhibited by FSLLRY-NH2. The protein level of P-P38MAPK , MDA contents and myocardial apoptosis rate increased and the activity of SOD decreased significantly (P<0.05 orP<0.01).ConclusionIschemic postconditioning has protective effect on myocardial ischemia reperfusion injury of aged diabetic rats. Its mechanism is related to the regulation of PAR-2/P38MAPK pathway.
Protease-activated receptor 2/P38MAPK ;Ischemic postconditioning; Ischemia reperfusion injury; Aged diabetic rats
10.3969/j.issn.1005-1740.2017.04.005
R541.4
A
1005-1740(2017)04-0024-05
湖北省自然科学基金(2014CKB1018)
华中科技大学同济医学院附属协和医院老年病科/老年医学研究所,武汉430022;#
,E-mail: yuji5832@163.com
本文2017-09-12收到,2017-10-26修回
