马克斯克鲁维酵母表达系统菊粉酶启动子的改造及活性研究
2017-12-14胡晓悦周峻岗
胡晓悦,周峻岗,2,余 垚,吕 红,2
(1. 复旦大学 生命科学学院 遗传工程国家重点实验室,上海 200438;2. 上海工业菌株工程技术研究中心,上海 200438)
马克斯克鲁维酵母表达系统菊粉酶启动子的改造及活性研究
胡晓悦1,周峻岗1,2,余 垚1,吕 红1,2
(1. 复旦大学 生命科学学院 遗传工程国家重点实验室,上海 200438;2. 上海工业菌株工程技术研究中心,上海 200438)
马克斯克鲁维酵母(Kluyveromycesmarxianus)作为一种食品安全级酵母,具有生长快,生物量高等特点,是一种新型的重组蛋白表达的宿主系统.启动子是供RNA聚合酶识别和结合的DNA序列,调控着转录的起始过程,直接影响基因的表达水平.本文通过易错PCR技术对马克斯克鲁维酵母菊粉酶启动子Pinu进行两轮突变,以阿魏酸酯酶(Est1E)作为报告基因,经筛选、鉴定获得了5个可以显著提高外源蛋白表达量的突变型启动子Pm1D81、Pm1F138、Pm2Q89、Pm2M73、Pm2X47,报告蛋白Est1E的酶活性是启动子改造前的2.31,2.65,5.01,5.40,5.43倍.随后测试了启动子Pm2X47对外源蛋白甘露聚糖酶和赤霉烯酮降解酶表达的影响,结果显示这两种酶的表达量均有提高,分别是启动子改造前的3.57倍和4.13倍.上述结果提示,改造后的菊粉酶启动子在提高外源蛋白表达水平方面具有一定的通用性,可作为新型候选表达元件,以提升马克斯克鲁维酵母重组表达外源蛋白的能力.
马克斯克鲁维酵母; 表达系统; 启动子; 优化改造
基因表达涉及转录、翻译及翻译后加工等多个方面.转录是基因表达的早期阶段,直接影响蛋白表达量的多少.转录效率受到转录元件的调控.转录元件包括启动子、终止子和增强子等,其中启动子是供RNA聚合酶特异性识别并结合的DNA序列,通过活化RNA聚合酶和相关转录因子,形成转录起始复合物,调控转录的起始过程.启动子的结构特征决定了它与RNA聚合酶的亲和力,进而影响目的基因的表达水平.
常见的启动子基本元件有: 转录起始位点(Transcription Start Site, TSS)、TATA框(TATA-box)、GGGCGG框(GC-box)、CAAT框(CAAT-box)等[1].转录起始位点是与RNA链第一个核苷酸相对应的DNA链上的碱基,通常为一个嘌呤(A或G),该处序列能够与转录因子特异性结合,促使DNA链在此解开并开始转录[2].TATA-box在真核生物中一般位于TSS上游25~35bp处,序列通常表现为TATAWTAW(W代表A或T).研究显示含有TATA-box的基因对转录调控更加敏感,其能够与RNA聚合酶和多种转录因子结合形成转录起始复合物(transcription Pre-Initiation Complex, PIC),调控转录起始[3-4].当PIC持续结合在TATA-box,能重复启动基因转录过程,从而提高基因的表达水平.CAAT-box一般位于TSS上游51~169bp处,其一致性序列为GGCTCAATCT.研究发现,CAAT-box对碱基突变十分敏感,一旦缺失或突变将导致转录效率急剧下降[5-6].除此之外,启动子上还存在些保守序列如GC-box和增强子.GC-box主要与转录起始频率有关,而增强子能够强化基因的转录起始,无方向性可位于基因的5’端和3’端.基于启动子结构信息,通过对启动子进行改造,筛选强启动子,是提高外源基因表达水平的一种重要方法[7].因此,利用易错PCR(Error Prone PCR, EP-PCR)技术对基因启动子区进行改造构建启动子文库,已经在微生物代谢工程中得到成功应用[8-10].Nevoigt等[11]通过筛选EP-PCR突变的溶氧敏感型启动子DAN1突变体文库,获得了在微氧条件下能够诱导表达的启动子突变体,其启动子活性在微氧条件下是野生型启动子的1.8~2.9倍.
马克斯克鲁维酵母(K.marxianus)是一种食品安全级酵母,它具有生长快、生物量高、耐高温、可利用多种碳源等特点,广泛应用于食品、医药和工业等领域[12-15].研究发现,K.marxianus可大量分泌菊粉酶,占胞外蛋白分泌量的50%以上[16].Bergkamp在该系统中利用菊粉酶启动子成功分泌表达α-半乳糖苷酶(alpha-galactosidase),表达量为153mg/L,而来源于酿酒酵母的强启动子PGK或GAL7,其表达量平均为2mg/L[17].本研究利用易错PCR技术对菊粉酶启动子进行突变改造,优化马克斯克鲁维酵母表达系统,提高重组蛋白表达能力,为分析基因转录调控提供理论依据.
1 材料与方法
1.1 材 料
1.1.1 菌株与质粒
马克斯克鲁维酵母尿嘧啶缺陷菌株KluyveromycesmarxianusFim1(ura3Δ),大肠杆菌E.coliDH5α、阿魏酸酯酶Est1E重组质粒表达载体pUKDN112-Est1E(本研究中命名为Pinu-Est1E)、赤霉烯酮降解酶ZHD101重组质粒表达载体pUKDN112-ZHD101(本研究中命名为Pinu-ZHD101)和甘露聚糖酶Mannase330重组质粒表达载体pUKDN112-Mannase330(本研究中命名为Pinu-Mannase330)均由本实验室保存和构建.
1.1.2 主要试剂
Phanta©Super-Fidelity DNA Polymerase(Vazyme公司),限制性内切酶、DNA连接酶、Carrier DNA(TaKaRa公司),GeneMorph II random mutagenesis kit(Agilent公司),胶回收试剂盒和质粒抽提试剂盒(Simgen公司),ZR Fungal/Bacterial RNA Mini PrepTM(ZYMO Research公司),PCR引物和DNA测序(上海杰李生物技术有限公司),多片段DNA一步酶连法[18]所需试剂来自New England Biolabs、TaKaRa、Inc和Epicentre.其他试剂均为国产分析纯.
1.1.3 培养基
LB培养基: 1%蛋白胨(Polypeptone),1%氯化钠(NaCl),0.5%酵母提取物(Yeast Extract),pH为7.0,氨苄抗性需加入100μg/mL氨苄西林(Amp);酵母完全培养基(YPD): 2%蛋白胨,1%酵母提取物,2%葡萄糖;SD-Uracil培养基: 0.67%YNB,2%葡萄糖,不含Uracil(尿嘧啶)的混合氨基酸;YD培养基: 2%酵母提取物,4%葡萄糖;若需要固体培养基,可在配制时加入2%琼脂糖.
1.2 方法
1.2.1 易错PCR扩增菊粉酶启动子Pinu片段
根据马克斯克鲁维酵母K.marxianus中菊粉酶序列(GenBank AB621573.1),其翻译起始密码子第一个碱基的前1216bp为启动子序列,设计启动子易错PCR扩增引物Pinu-F(5’-CGCTGCAACGGCATGCCGA-TCGAAAAGGTAAAC-3’)和Pinu-R(5’-AGTCCCGGGGTCACCGTCTCTCTTGTAATTGATC-3’).加样按GeneMorph II random mutagenesis kit(Agilent公司)说明书进行,突变频率为每100~500ng模板,其1kb内碱基的突变个数为4.5~9个.每50μL反应体系为: 5μL 10× MutazymeII reaction buffer;1μL 40mmol/L dNTP Mix;1U MutazymeII DNA polymerase;引物Pinu-F和Pinu-R各15pmol;质粒模板Pinu-Est1E为150~200ng,加无菌水补足至50μL.反应程序为: 95℃ 3min;95℃ 30s,56℃ 30s,72℃ 1min 20s,30个循环;72℃ 5min.反应完成后用1%的琼脂糖凝胶电泳鉴定并回收产物.
1.2.2 启动子突变体文库的构建
采用特长引物全质粒扩增法(Mega-WHOP)[19-20],以随机突变后的目标基因Pinu作为双链互补的引物对,表达载体Pinu-Est1E作为模板(此模板质粒甲基化),采用高保真DNA聚合酶进行PCR,扩增出质粒的全长片段;再用DpnⅠ酶处理甲基化质粒模板,新扩增出的质粒因为没有甲基化而被保留.每50μL反应体系为: 25μL 2× Phanta Max Buffer;1.5μL 10mmol/L dNTP Mix;PCR产物Pinu大于600ng;质粒模板Pinu-Est1E大于100ng;1.5U Phanta Max Super-Fidelity DNA Polymerase,加无菌水补足至50μL.反应程序为: 95℃ 5min;95℃ 50s,60℃ 50s,72℃ 10min,30个循环;72℃ 10min.反应完成后,加入1UDpnⅠ,37℃ 2h,消化模板.产物转化至E.coliDH5α细胞,涂布LB(含100μg/mL Amp)平板,37℃过夜培养,挑单克隆至96孔细胞培养板中(每个孔对应一个样本),送公司进行质粒抽提,然后采用96孔板酵母化学转化方法(将Fim1接入到50mL YPD培养基中,30℃,220r/min培养过夜,次日将过夜菌转移至50mL离心管中,全部离心8000r/min 10min,弃上清.用无菌水洗涤菌体一次,8000r/min 10min弃上清;1× LiAc/TE洗两遍,8000r/min 10min弃上清;先用排枪吸取5μL已抽提质粒加入96孔板中,然后在处理好的菌体中加入200μL载体DNA、25mL PEG溶液和20μL 1mol/L DTT.充分混匀后,排枪吸取200μL混合液加入96孔板中,30℃ 15min,47℃ 15min后,3700r/min 3min离心,弃上清,100μL无菌水悬浮菌体,然后用钉板将菌体涂布SD-Ura固体平板,30℃倒置培养48h.),转化至Fim1细胞,用钉板打点在SD-Ura固体培养基平板上,经平板筛选出的阳性克隆作为突变株进行后续筛选.菊粉酶启动子部分测序引物为Pinu-CX-R1(5’-GATTCGCACAGAAACGCCGGATGAC-3’)和Pinu-CX-R2(5’-CTATGTCCTGTAAAGCCATGAATGATG-3’)
1.2.3 高效启动子重组菌株筛选方法
质粒Pinu-Est1E的报告基因是阿魏酸酯酶,以重组菌在YD培养基中发酵72h后Est1E的酶活作为检测对象,比较启动子突变型菌株与野生型菌株的Est1E酶活.初筛挑单克隆于含600μLYD培养基的24孔板中进行发酵,复筛挑单克隆于含50mL YD培养基的150mL摇瓶进行发酵.实验选用阿魏酸-2-氯-对硝基苯酚酯(CNPF)作为阿魏酸酯酶高通量筛选体系的反应底物,它可以真实反映Est1E的酶活,且410nm处吸光值与Est1E酶活呈剂量依赖型,即能够体现各样本间酶活的差异[21-22].每200μL反应体系: 20μL发酵液上清,10μL 20mmol/L CNPF,170μL 1×PBST(1×PBS Buffer pH 6.4;2.5% Triton X-100);37℃ 25min.阿魏酸酯酶相对酶活=(样本OD410-阴性对照OD410)/(野生型OD410-阴性对照OD410).
1.2.4 Real-Time PCR检测报告基因的转录水平
将菌体接种于含YD的试管中过夜培养,第二天转接至150mL摇瓶中,起始OD600=0.1,生长至对数期OD600=0.6~0.8时收菌.用ZR Fungal/Bacterial RNA Mini PrepTM(ZYMO Research)试剂盒抽提RNA,利用反转试剂盒(TaKaRa)进行RT-PCR反应,随后采用SYBR premix Ex-Taq(TaKaRa)和LightCycler480(Roche)进行定量PCR分析,以KmActin作为内参,RT-PCR引物序列参见表1.
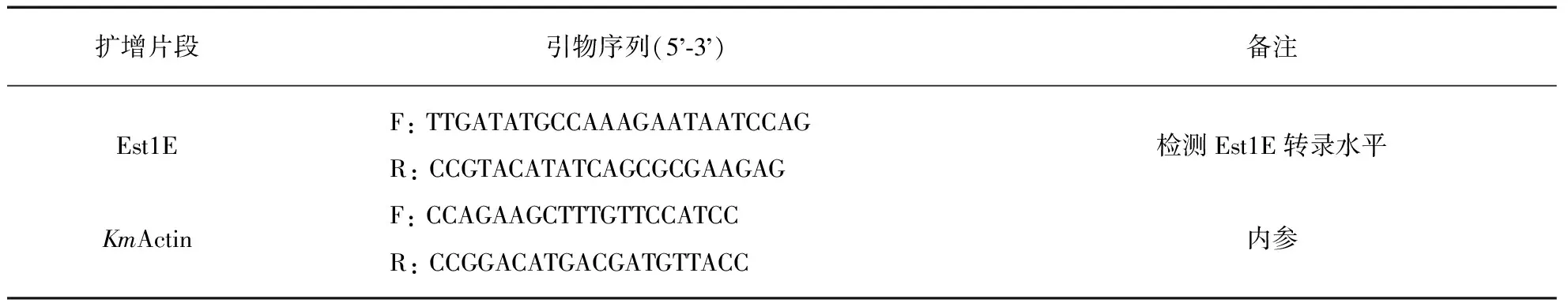
表1 RT-PCR引物序列Tab.1 The sequence of RT-PCR primers
1.2.5 高效启动子通用性检测质粒的构建
启动子筛选是以报告基因Est1E相对酶活的高低作为筛选依据,为检测高效突变型启动子是否适用于其他外源蛋白,将启动子后报告基因替换为甘露聚糖酶(Mannase330)和赤霉烯酮降解酶(ZHD101).用SmaⅠ和NotⅠ双酶切含有高效突变型启动子的质粒,回收大片段.同时用高保真酶以Pinu-Mannase330和Pinu-ZHD101为模板,克隆两种外源基因Mannase330和ZHD101并回收,采用一步法重组技术[18]将质粒大片段和目的基因连接起来.外源基因扩增及测序引物序列均为N112-Gene-F(5’-CA-GTGATCAATTACAAGAGAGACGGTGAC-3’)和N112-Gene-R(5’-GTAAGCAGATCAGATCAAA-GCTTGCGGCC-3’).
1.2.6 重组菌中甘露聚糖酶和赤霉烯酮降解酶表达分析
挑鉴定后的阳性重组菌单克隆于50mL YPD培养基中,30℃,220r/min,培养72h,吸取发酵上清液检测酶活.
甘露聚糖酶(Mannase330)酶活检测方法——DNS法[23]: 5μL发酵上清液与145μL 0.5%槐豆胶溶液混合,70℃反应10min,立即加入150μL DNS,沸水浴反应10min,冷却后在570nm处测定吸光度.Mannase330相对酶活=(样本OD570-阴性对照OD570)/(野生型OD570-阴性对照OD570).
赤霉烯酮降解酶(ZHD101)酶活检测方法——高效液相色谱法分析(HPLC): 10μL发酵上清液与90μL 50ng/μL ZEN底物混合,37℃反应30min,立即加入100μL乙腈,4℃ 14000r/min离心15min,取上清液进行HPLC检测.HPLC检测选用Agilent XDB-C18色谱柱(250mm×4.6mm,5μm),检测波长为240nm,色谱流动相条件: 0~8min(甲醇20%~80%);8~12min(甲醇80%);12~16min(甲醇80%~20%);流速为1mL/min.ZHD101相对酶活=(阴性对照峰面积-样本峰面积)/(阴性对照峰面积-野生型样本峰面积).
2 结 果
2.1 启动子Pinu突变改造及突变体文库的构建
利用Agilent随机突变试剂盒,以含有菊粉酶启动子序列的载体Pinu-Est1E为模板,模板量为135ng,对全长1216bp的菊粉酶启动子进行易错PCR扩增,PCR结果如图1(a)所示.易错PCR产物纯化后作为引物,用PCR方法扩增表达载体Pinu-Est1E全序列,产物经DpnⅠ酶消化处理后,转化大肠杆菌E.coliDH5α感受态细胞.随机挑取10个克隆对启动子段进行测序,发现每1kb平均有7个碱基发生突变,达到构建突变体文库的要求.抽提大肠杆菌转化子质粒,用化学转化法转入酵母Fim1细胞,所有转化子构成突变体文库(图1(b)).
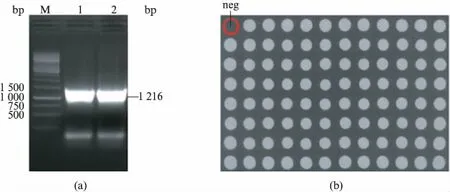
图1 Pinu突变体文库在Fim1酵母细胞中的构建Fig.1 Construction of Pinu mutant library in Fim1(a) 易错PCR扩增菊粉酶启动子,M: 1kb DNA marker;1,2: 菊粉酶启动子易错PCR扩增产物;(b) 在96孔板中用化学方法转化Fim1酵母细胞,构建启动子突变体文库,第一个为阴性对照.
2.2 高效菊粉酶启动子突变体的筛选
为得到高效的启动子Pinu,以外源蛋白Est1E的表达作为报告蛋白,以其酶活高低作为筛选依据.本研究对启动子Pinu进行两轮易错PCR,在第一轮易错PCR中,以野生型启动子Pinu作为模板,在Fim1酵母细胞中构建了第一轮的启动子突变体文库,并筛选了3500个突变体,分析了突变株中Est1E相对于含野生型菊粉酶启动子菌株(WT)中Est1E的表达水平(图2(a),见第442页).经初筛和复筛,获得2个阿魏酸酯酶酶活相对较高的突变株D81和F138,其表达量是野生型启动子介导的Est1E表达量的2.31,2.65倍(图2(c)).
为进一步得到更强启动效率的启动子,以D81和F138含有的突变启动子作为混合模板,再次用易错PCR扩增启动子片段,并在Fim1酵母菌株中构建了第二轮易错PCR的启动子突变体文库,筛选了3000个突变体,报告蛋白Est1E的相对表达量如图2(b)所示.经初复筛后,最终获得3个Est1E酶活相对较高的突变株Q89、M73、X47,其表达量分别是野生型启动子介导作用下的5.01,5.40,5.43倍(图2(c)).
同时,利用RT-PCR对高效表达菌株中的Est1E转录水平进行了分析,结果发现突变株D81、F138、Q89、M73、X47的Est1E的转录水平较野生型均有很大的提高,分别是野生型的3.8,5.0,5.6,17.7,35.6倍(图2(d)).说明启动子序列发生的突变,提高了外源基因的转录水平,从而使蛋白的分泌表达量提高.
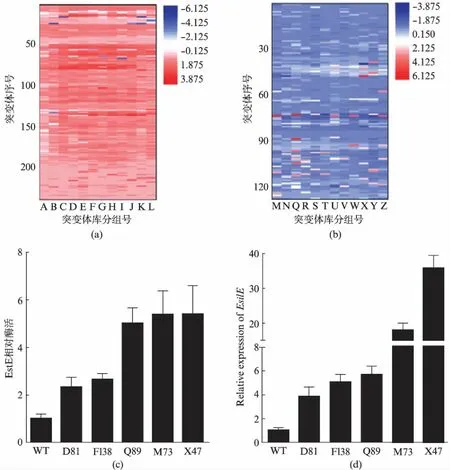
图2 高效菊粉酶启动子的筛选Fig.2 Screening of highly efficient Inulinase promoter(a) 第一轮易错PCR的Pinu突变体库筛选,热谱图表示了Pinu改造后Est1E酶活变化情况;(b) 第二轮易错PCR的Pinu突变体库筛选,热谱图表示了Pinu第二次改造后Est1E酶活变化情况;(c) 启动子突变对Est1E酶活水平的影响;(d) 启动子突变对Est1E转录的影响.
2.3 高效启动子序列分析
通过两轮易错PCR改造和筛选,共获得了5个阿魏酸酯酶表达量较高的菌株,其对应的启动子分别为Pm1D81、Pm1F138、Pm2Q89、Pm2M73、Pm2X47.利用引物Pinu-CX-R1和Pinu-CX-R2对上述启动子进行测序,并与原始的启动子序列(菊粉酶序列GenBankAB621573.1,启动子范围为331~1546)进行对比,突变体碱基变化如表2所示.结果表明,利用易错PCR技术成功的在Pinu中引入突变,第一轮易错PCR改造后,获得高效突变型启动子Pm1D81和Pm1F138,突变位点随机分布,碱基突变率为0.53%.启动子Pm2Q89、Pm2M73、Pm2X47是在突变启动子Pm1D81和Pm1F138基础上进行第二轮易错PCR扩增得到的,其中Pm2Q89有4个突变点与Pm1F138相同,却屏蔽了第951位的点突变;Pm2M73、Pm2X47既兼具了Pm1F138的突变位点,同时也引入了新的突变点,其中Pm2X47有部分突变位点与Pm1D81相同.从总体测序结果来看,突变位点主要集中在启动子序列的1~200bp、600~800bp、900~1200bp区域内.
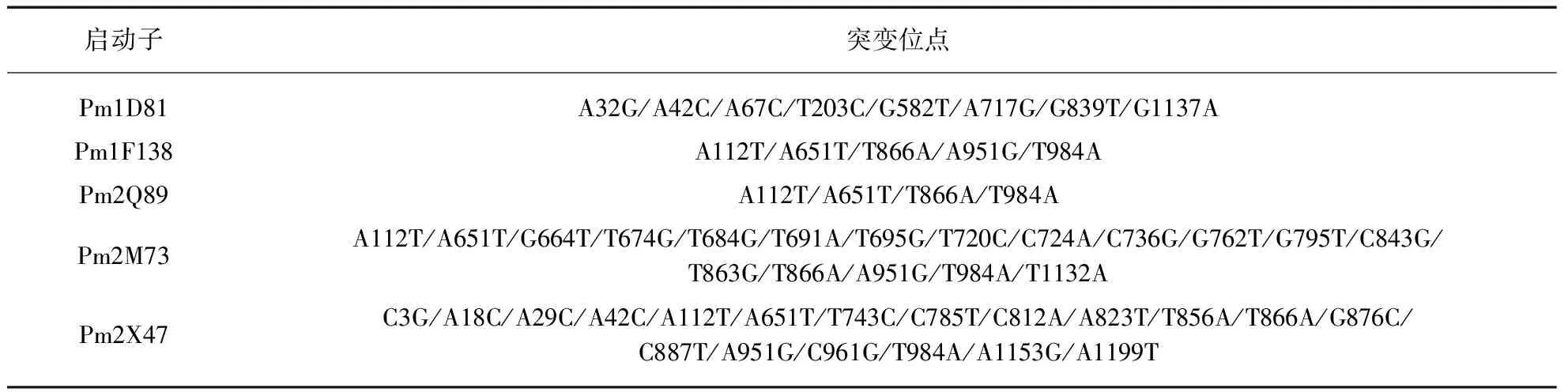
表2 突变型启动子的突变位点Tab.2 The mutation sites of mutant promoters
注: 菊粉酶启动子全长1216bp,将第一个碱基A记为+1,表中A32G表示启动子第32位碱基A突变为碱基G.
2.4 高效启动子通用性检测
为检测突变后的启动子是否可以提高其他蛋白的表达量,我们抽提了阿魏酸酯酶表达量最高菌株X47的质粒,用SmaⅠ和NotⅠ酶切,回收不含有Est1E编码基因的质粒载体大片段,随后分别将甘露聚糖酶编码基因Mannase330(GenBank JQ796716.1)和赤霉烯酮降解酶编码基因zhd101(GenBank KR363960.1)克隆到回收的载体大片段上,构建获得重组质粒Pm2X47-Mannase330和Pm2X47-ZHD101.测序正确后将质粒Pm2X47-Mannase330和Pm2X47-ZHD101分别转入Fim1菌株中,挑取单克隆接种摇瓶,发酵72h后,取上清分别检测酶活.结果显示,在突变启动子Pm2X47的介导下,Mannase330和ZHD101的酶活明显高于野生型启动子Pinu介导下表达的酶活(图3(a)),分别是野生型的3.57和4.13倍.
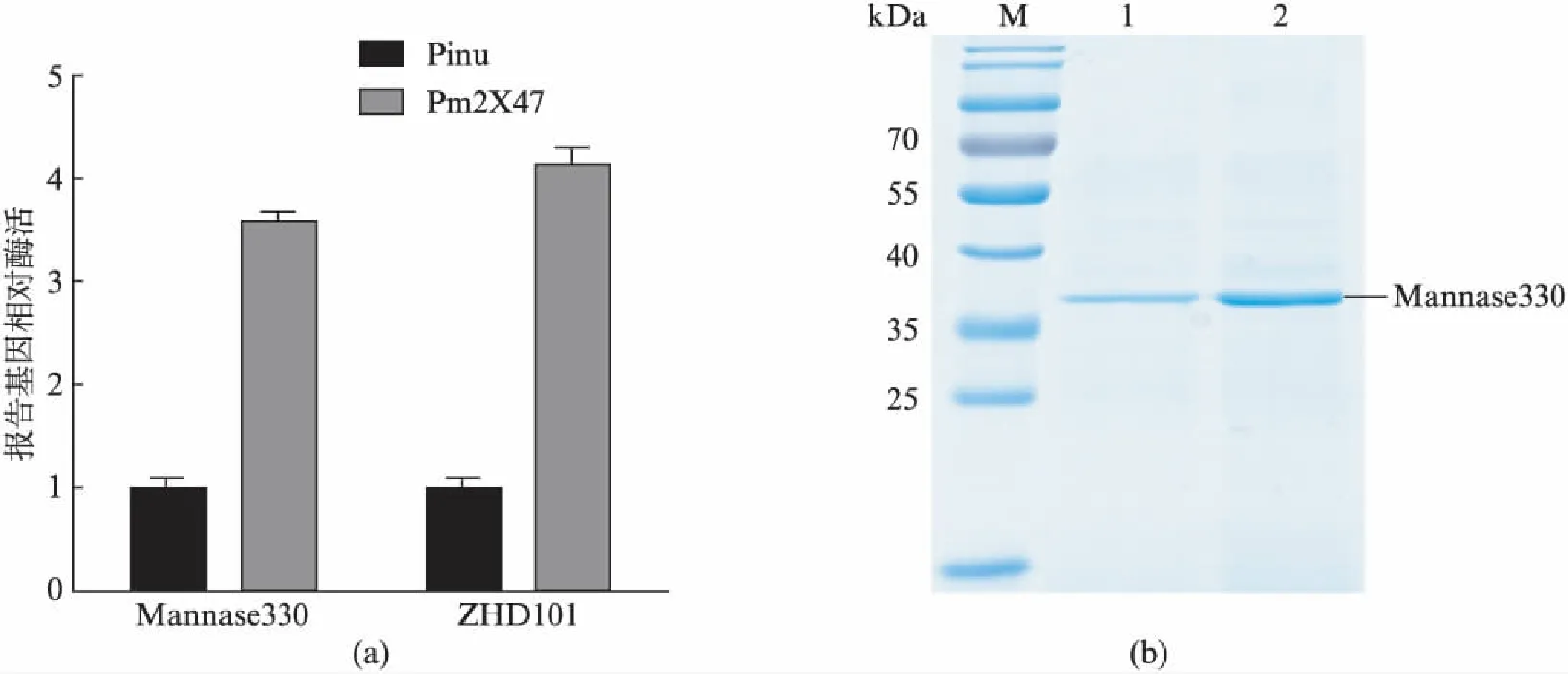
图3 高效启动子通用性研究Fig.3 Study on the universality of highly efficient promoter(a) 突变启动子Pm2X47对不同外源蛋白表达水平提高的影响;(b) 甘露聚糖酶野生型菌株和重组菌株发酵液上清SDS-PEG,M: pageruler prestained protein ladder;1. Fim1/Pinu-Mannase330发酵液上清;2. Fim1/Pm2X47-Mannase330发酵液上清.
同时,对Fim1/Pinu-Mannase330和Fim1/Pm2X47-Mannase330表达的甘露聚糖酶进行SDS-PEG电泳检测.从蛋白质凝胶电泳的结果来看,两株重组菌都能分泌表达甘露聚糖酶,38kDa处蛋白条带明显,且与甘露聚糖酶理论分子量一致.同时,我们也发现Fim1/Pm2X47-Mannase330菌株甘露聚糖酶的表达量明显高于Fim1/Pinu-Mannase330(图3(b)).这一结果说明,通过易错PCR获得的突变启动子Pm2X47能够很好地提高外源蛋白的表达水平,突变启动子Pm2X47具有更高的转录效率;同时这种高效的突变启动子也拥有一定的普适性,具有提高多种外源蛋白表达水平的能力,为马克斯克鲁维酵母表达系统的优化提供了高效表达元件.
3 讨 论
蛋白质的高效表达一直是生物研究和工业应用的热点.蛋白质高效表达系统主要包含两个方面: 宿主和载体.马克斯克鲁维酵母比毕赤氏酵母、酿酒酵母、乳酸克鲁维酵母等的生长速率快,同时具有高生物量、耐高温、可利用多种碳源等优良特性,因此在重组蛋白的工业生产中具有广阔的应用前景[24-27].调节外源蛋白表达的核心元件由启动子、外源基因、信号肽和终止子等构成,外源基因在细胞内的表达水平与基因的转录强度有密切的关系.启动子作为转录调控的重要元件,在相关转录因子的作用下与RNA聚合酶识别并结合,并指导转录的开始.启动子区的结构影响其与RNA聚合酶的结合能力,进而影响基因的转录表达效率.
本研究为获得效率更高的菊粉酶启动子以提高外源蛋白的表达水平,利用易错PCR技术,在原始Pinu序列中引入随机突变,以Est1E作为报告基因.经过两轮的易错PCR改造,最终获得了5个高效启动子Pm1D81、Pm1F138、Pm2Q89、Pm2M73、Pm2X47,Est1E的酶活分别是启动子改造前的2.31、2.65、5.01、5.40、5.43倍.经酶活检测和测序发现,第二轮易错PCR改造后产生的有益突变并不如第一轮的多,并且每一轮改造后产生的突变位点随机,突变频率也相对较高.两轮易错PCR的反应条件及程序虽完全一致,但在相同条件下也可能存在偏好突变位点的情况.真核启动子有些保守区域如GC-box,该区域碱基的变化可以控制转录水平的高低[5-6].菊粉酶启动子的GC-box在+621~+838bp范围内,5个突变型启动子在这些区域均有突变产生,且Est1E酶活上升,可以认为是该区域的点突变提高了启动子的活性,促进了报告基因的转录效率.同时,我们也发现启动子Pm1F138、Pm2Q89、Pm2M73、Pm2X47在+112bp处均存在碱基A变为T,形成了一个TATA-box序列(TATATTA).有研究发现,当转录起始复合物(PIC)持续结合在TATA-box上时,可以使转录重复进行[3-4].上述4种启动子对转录过程的促进作用可能受该处点突变形成的TATA-box与PIC重复结合作用的影响.由于启动子序列上的点突变相对较为分散且规律性不强,其对转录的促进作用很有可能是各处点突变及其相关转录因子共同作用的结果,具体的机制有待进一步实验确认.
本研究中,我们发现易错PCR作为基因工程改造技术可以有效地优化启动子,并且获得的高效启动子具有一定的普适性.我们用高效启动子Pm2X47介导甘露聚糖酶和赤霉烯酮降解酶在Fim1菌株中表达,发现与野生型启动子Pinu相比,两种酶的表达量都有提高,分别是野生型启动子介导作用下的3.57和4.13倍.这为高效菊粉酶启动子适用于其他异源蛋白高效表达提供了强有力的理论依据.通过对菊粉酶启动子进行改造,马克斯克鲁维酵母表达系统在外源基因表达上有了提升,为该系统在食品医药及工业等领域的应用奠定了良好的基础.
[1] BARROSO-DEL JESUS A, ROMERO-LOPEZ C, LUCENA-AGUILAR G,etal. Embryonic stem cell-specific miR302-367 cluster: Human gene structure and functional characterization of its core promoter [J].MolCellBiol, 2008,28(21): 6609-6619.
[2] DING W, BELLUSCI S, SHI W,etal. Genomic structure and promoter characterization of the human Sprouty4 gene, a novel regulator of lung morphogenesis [J].AmJPhysiolLungCellMolphysiol, 2004,287(1): 52-59.
[3] EMOTO M, MIKI M, SARKER AH,etal. Structure and transcription promoter activity of mouse flap endonuclease1gene: Alternative splicing and bidirectional promoter [J].Gene, 2005,357(1): 47-54.
[4] 张小辉,祁艳霞.真核生物启动子TATA-box, GC-box和CAGT-box的分析 [J].安徽农业科学,2008,36(4): 1380-1381.
[5] HERNANDEZ-RODRIGUEZ CS, FERRE J, HERRERO S. Genomic structure and promoter analysis of pathogen-induced repat genes fromSpodopteraexigua[J].InsectMolBiol, 2009,18(1): 77-85.
[6] KHARITONOVA MA, VERSHININA VI, MOROZOVA OV,etal. The role of the promoter structure in the efficiency of the expression of guanylspecific ribonucleases fromBacillusintermediusandBacilluspumilus [J].MolGenMikrobiolVirusol, 2006(4): 15-19.
[7] ALPER H, FISCHER C, NEVOIGT E,etal. Tuning genetic control through promoter engineering [J].ProcNatlAcadSciUSA, 2005,102(36): 12678-12683.
[8] BRAATSCH S, HELMARK S, KRANZ H,etal.Escherichiacolistrains with promoter libraries constructed by Red/ET recombination pave the way for transcriptional fine-tuning [J].Biotechniques, 2008,45(3): 335-337.
[9] SIEGL T, TOKOVENKO B, MYRONOVSKYI M,etal. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes [J].MetabolicEngineering, 2013,19: 98-106.
[10] RYTTER JV, HELMARK S, CHEN J,etal. Synthetic promoter libraries forCorynebacteriumglutamicum[J].AppliedMicrobiologyandBiotechnology, 2014,98(6): 2617-2623.
[11] NEVOIGT E, FISCHER C, MUCHA O,etal. Engineering promoter regulation [J].BiotechnologyandBioengineering, 2007,96(3): 550-558.
[12] FONSECA G G, HEINZLE E, WITTMANN C,etal. The yeastKluyveromycesmarxianusand its biotechnological potential [J].AppliedMicrobiologyandBiotechnology, 2008,79(3): 339-354.
[13] URIT T, LÖSER C, WUNDERLICH M,etal. Formation of ethyl acetate byKluyveromycesmarxianuson whey: Studies of the ester stripping [J].BioprocessBiosysteng, 2011,34(5): 547-559.
[14] LANE M M, MORRISSEY J P.Kluyveromycesmarxianus: A yeast emerging from its sister’s shadow [J].FungalBiolRev, 2010,24(1/2): 17-26.
[15] RODICIO R, HEINISCH J J. Yeast on the milk way: Genetics, physiology and biotechnology ofKluyveromyceslactis[J].Yeast, 2013,30(5): 165-177.
[16] KANGO N, JAIN SC. Production and properties of microbial inulinases: Recent advances [J].FoodBiotechnol2011,25: 165-212.
[17] BERGKAMP RJ, BOOTSMAN TC, TOSCHKA HY,etal. Expression of an alpha-galactosidase gene under control of the homologous inulinase promoter inKluyveromycesmarxianus[J].MicrobiolBiotechnol, 1993,40(2/3): 309-317.
[18] GIBSON D G, YOUNG L, CHUANG R Y,etal. Enzymatic assembly of DNA molecules up to several hundred kilobases [J].NatureMethods, 2009,6(5): 343-345.
[19] MIYAZAKI K, TAKENOUCHI M. Creating random mutagenesis libraries using megaprimer PCR of whole plasmid [J].Biotechniques, 2002,33(5): 1033-1038.
[20] WEI D, LI M, ZHANG X,etal. An improvement of the site-directed mutagenesis method by combination of megaprimer, one-side PCR andDpnⅠ treatment [J].AnalyticalBiochemistry, 2004,331(2): 401-403.
[21] HEGDE S, SRINIVAS P, MURALIKRISHNA G. Single-step synthesis of 4-nitrophenyl ferulate for spectrophotometric assay of feruloy lesterases [J].AnalyticalBiochemistry, 2009,387(1): 128-129.
[22] ZHANG S, MA X, PEI X,etal. A practical high-throughput screening system for feruloyl esterases: Substrate design and evaluation [J].JournalofMolecularCatalysisB:Enzymatic, 2012,4(1/2): 36-40.
[23] MILLER G L. Use of Dinitrosalicylic acid reagent for determination of reducing sugar [J].AnalChem, 1959,31(3): 426-428.
[24] WEN T, LIU F, HUO K,etal. Cloning and analysis of the inulinase gene fromKluyveromycescicerisporusCBS4857 [J].WorldJournalofMicrobiologyandBiotechnology, 2003,19(4): 423-426.
[25] ZHANG J, YUAN H, WEN T,etal. Cloning of theKcURA3 gene and development of a transformation system forKluyveromycescicerisporus[J].ApplMicrobiolBiotechnol, 2003,62(4): 387-391.
[26] KONDO K, MIURA Y, SONE H. High-level expression of a sweet protein, monellin, in the food yeastCandidautilis[J].NatBiotechnol, 1997,15(5): 453-457.
[27] CAI X P, ZHANG J, YUAN H Y,etal. Secretory expression of heterologous protein inKluyveromycescicerisporus[J].ApplMicrobiolBiotechnol, 2005,67(3): 364-369.
[28] WESTHOLM J O, XU F, RONNE H,etal. Genome-scale study of the importance of binding site context for transcription factor binding and gene regulation [J].BMCBioinformatics, 2008(9): 484-498.
StudyontheTransformationandActivityoftheInulinasePromoterinKluyveromycesmarxianus
HUXiaoyue,HUXiaoyu1,2,ZHOUJungang1,2,YUyao1,LÜHong1,2
(1.StateKeyLaboratoryofGeneticEngineering,SchoolofLifeSciences,FudanUniversity,Shanghai, 200438,China;2.ShanghaiEngineeringResearchCenterofIndustrialMicroorganisms,Shanghai, 200438,China)
As a food safety grade yeast,Kluyveromycesmarxianusis a new host system for recombinant protein expression, which has the characteristics of fast growth and high biomass. The promoter is a DNA sequence which is recognized and bound by RNA polymerase that regulates the initiation of transcription and gene expression level. In this study, two rounds of random mutagenesis of the inulinase promoter have been carried out by the error-prone PCR technique. Finally, we got five highly efficient mutant promoters by screening and identification of the expression level of ferulic acid esterase(Est1E). The results showed that the mutant promoters Pm1D81, Pm1F138, Pm2Q89, Pm2M73, Pm2X47 can improve the reporter protein Est1E activity, which were 2.31,2.65,5.01,5.40,5.43 times as much as that of the wild-type promoter. The effect of promoter Pm2X47 on the expression of other exogenous proteins mannanase and zearalenone-degrading enzyme has been tested. The results showed that the Pm2X47-mediated expression of two enzymes were both significantly improved, respectively, 3.57 times and 4.13 times before the transformation of the inulinase promoter. These results indicate that the modified inulinase promoter has a certain universality in improving the expression level of exogenous protein and provides a novel candidate expression element for enhancing the ability of expression of recombinant protein inKluyveromycesmarxianusexpression system.
Kluyveromycesmarxianus; expression system; promoter; transformation and optimization
0427-7104(2017)04-0438-08
2017-01-25
国家高技术研究发展计划项目(2013AA102803B,2014AA021301);上海市科委基地项目(13DZ2252000)
胡晓悦(1992—),女,硕士研究生;吕 红,女,教授,通信联系人,E-mail: honglv@fudan.edu.cn.
Q814
A
