正十六烷纳米液滴在光滑壁面上润湿行为的分子动力学模拟
2017-12-01,,
, ,
(大连理工大学 化工学院 , 辽宁 大连 116024)
正十六烷纳米液滴在光滑壁面上润湿行为的分子动力学模拟
白麟,王宝和,于志家
(大连理工大学 化工学院 , 辽宁 大连 116024)
采用分子动力学模拟技术,研究了正十六烷纳米液滴在光滑壁面上的润湿行为规律。模拟结果表明,壁面厚度、长度(或宽度)、截断半径及分子数对接触角的影响不大。随着壁面作用势能的增大,接触角线性减小;当壁面作用势能为0.5 kJ/mol时,接触角约为90°。随着模拟温度的提高,接触角逐渐减小。
分子动力学 ; 模拟 ; 接触角 ; 正十六烷
在现代社会中,经济的高速发展可能会产生大量被有机物污染的工业水资源;同样,石油的开采、运输以及存储过程中,均易发生油品泄漏等污染事件[1-2]。与此同时,航空齿轮的润滑和微/纳机电系统的冷却都与液滴的润湿性有关。此外,航空齿轮润滑油的铺展特性极大地影响着轴承腔的润滑与散热功能,因此,开展油滴成膜的流动铺展特性研究,为机械零部件润滑计算提供准确合理的基础数据,对于实现飞行器中的机械零部件精确润滑设计十分重要[3]。为了更好地理解液滴润湿铺展行为机理,越来越多的研究学者将重点放在分子水平层次上。近年来,随着计算机技术的迅猛发展,也使分子动力学模拟技术计算纳米液滴的润湿性质成为可能。Werder等[4]利用分子动力学模拟了石墨表面上水液滴的接触角与壁面作用势能之间的作用关系。邱丰等[5]采用分子动力学模拟的方法,对Pb液滴在Cu基底上的铺展润湿行为进行研究,发现晶体结构对液滴铺展具有较大影响。但目前还没有关于烷烃类纳米液滴润湿性的分子动力学模拟研究的报道。本文将采用分子动力学模拟的方法,利用LAMMPS软件模拟光滑壁面上正十六烷纳米液滴的润湿性质,从壁面厚度和宽度(或长度)、分子数、壁面作用势能及模拟温度等方面,对接触角的影响进行探究。
1 模拟方法
1.1模拟体系的建立
模拟体系的初始构型如图 1所示,壁面采用面心立方排布的Cu原子,晶格长度为0.361 5 nm,总共306 545个Cu原子,模拟盒子尺寸为47 nm×47 nm×20 nm。2 000个(研究分子数影响时除外)正十六烷分子随机分布成球形液滴,正十六烷分子的初速度由随机数发生器确定[6]。液滴质心与壁面之间的初始距离为6.447 nm。

图1 初始模型
1.2势能模型的选取
当所研究的粒子中含有的原子数目较多时,通常采用的原子模型包括全原子模型和联合原子模型两种形式。在全原子模型中,将体系中的每个原子看作一个基本单元,由Material Studio软件得到如图 2所示的正十六烷分子结构,其中灰色表示为C原子,白色表示H原子,并且在计算过程中定义正十六烷上每一个原子的参数,包括侧链烷基上的H原子,这样的力场称为全原子力场。在分子动力学模拟中,为了简化计算,有时将一些原子团如CH2被看作一个原子,称作虚拟原子,如图 3所示,黑色的CH3和灰色的CH2被当作相对分子质量为15和14虚拟原子,这样简化的力场称作联合原子力场。
为了简化计算,本模拟采用联合原子力场模型。

图2 全原子模型

图3联合原子模型
正十六烷分子间的势能函数如式(1)所示[7]。

(1)
式中:U(tot)为总势能;U(NB)和U(B)分别为非键势能及键合势能;N为正十六烷分子数;rij为i分子和j分子中两个虚拟原子之间的距离;σij为i和j分子中两个虚拟原子之间L-J势能的尺度参数;εij为i和j分子中两个虚拟原子之间L-J势能的能量参数;kr和kθ分别为键长伸缩弹力系数和键角弯曲弹力系数;k1~k4为二面角扭转势能弹力系数;r、θ分别为键长、键角;r0、θ0分别为平衡键长、键角;φ为二面角。键长、键角、二面角作用参数如表1~3所示[8]。

表1 键长作用参数

表2 键角作用参数

表3 二面角作用参数 kJ/mol
不同虚拟原子间或固体壁面原子与虚拟原子间的势能函数仍为式(1),其L-J 势能的能量参数和尺度参数采用混合规则计算,如式(2)和(3)所示[9]。

(2)

(3)
式中:εls为虚拟原子与固体壁面原子之间L-J势能的能量参数;εll为相同虚拟原子之间L-J 势能的能量参数;εss为壁面原子之间L-J 势能的能量参数。σls为虚拟原子与壁面原子之间L-J势能的长度参数;σll为相同虚拟原子之间L-J势能的长度参数;σss为壁面原子之间L-J势能的长度参数(σss=0.234 nm)。虚拟原子L-J势能参数如表4所示[8]。

表4 虚拟原子L-J势能参数
1.3模拟细节
模拟在x、y方向采用周期性边界条件,在z方向采用固壁和镜像边界条件;粒子间力的截断半径为1.367 nm,模拟时间步长为1 fs,总模拟时间为2 ns,前1 ns使得系统达到平衡,后1 ns统计计算并输出系统的密度分布。采用正则系综(NVT),并用Woodcock控温法维持体系温度衡定;每隔1 000步矫正体系的质心。模拟数据采用LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)软件计算得到。采用等密度拟合曲线法计算液滴密度和接触角[10]。
2 结果与讨论
2.1模拟参数的影响
2.1.1壁面厚度的影响
选择47.00 nm×47.00 nm×20.00 nm的模拟盒子,正十六烷分子数为2 000,截断半径为1.367 nm,壁面作用势能为1.5 kJ/mol,温度为298 K,当壁面厚度D分别为1.084、1.446、1.807、2.169 nm时,进行计算模拟,得到图 4所示的一维密度分布(D=1.446 nm,其他壁面厚度的类似)和图5所示的二维密度图(D=1.446 nm,其他壁面厚度的类似),其中图5中颜色灰度从浅到深代表密度从ρ=0到ρ=1.250 g/cm3。根据二维密度图,再采用文献[10]的方法,得到的接触角如图6所示。
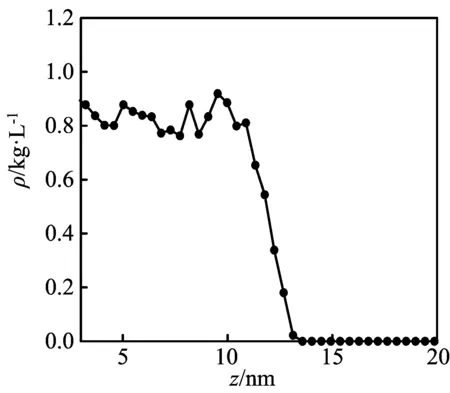
图4 正十六烷纳米液滴的一维密度图(D=1.446 nm)

图5 正十六烷纳米液滴的二维密度图(D=1.446 nm)

图6 壁面厚度(D)对接触角(θ)的影响
由图 4可见,液体主体密度有小幅度波动,统计平均值为0.799 2 kg/L,接近于常温下正十六烷液体密度的实验值(0.78 kg/L)[11]。对于壁面作用势能1.5 kJ/mol的光滑壁面,当壁面厚度大于分子间作用力的截断半径时,不同壁面厚度下模拟得到的接触角基本相同,约为30°,即壁面厚度对接触角影响不大。因此,本文的模拟研究,其壁面厚度均采用1.446 nm。
2.1.2壁面宽(或长)度的影响
选择盒子高度为20.00 nm,壁面厚度为1.446 nm,正十六烷分子数为2 000,截断半径为1.367 nm,壁面作用势能为1.5 kJ/mol,温度为298 K,壁面宽(或长)度即盒子宽(或长)度L分别为36.15、39.77、43.38、47.00、50.61 nm时,进行模拟计算,可以得到正十六烷纳米液滴的二维密度分布图,根据二维密度图,再采用文献[10]的方法,计算得到的接触角如图7所示。
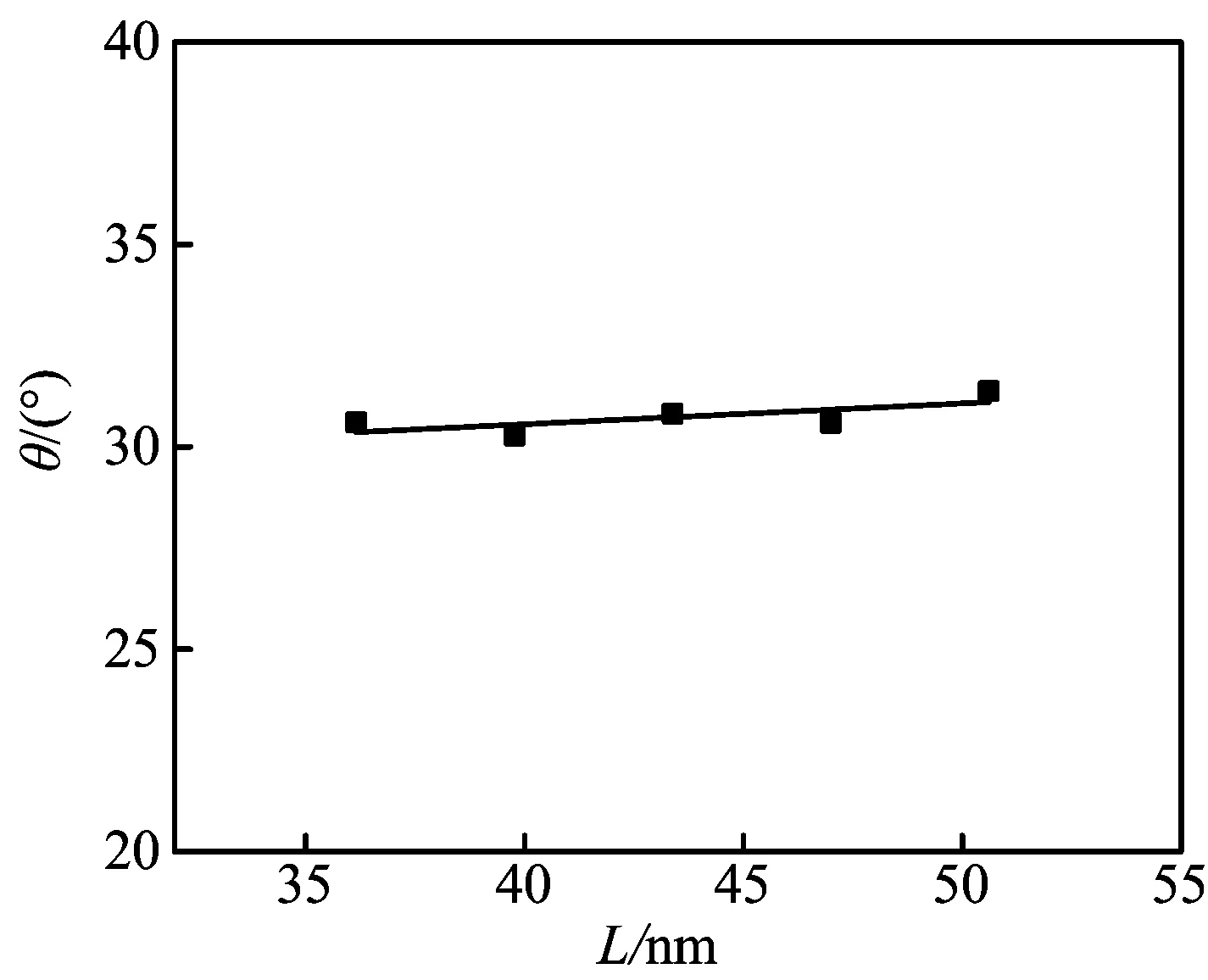
图7 壁面长度(或宽度)(L)对接触角(θ)的影响
由图 7可以看出,截断半径小于模拟盒子的一半时,模拟得到的接触角均为30°左右,即固体壁面宽(或长)度对接触角影响不大。本文的模拟研究,其壁面宽(或长)度均采用47.00 nm。
2.1.3分子数的影响
将模拟盒子设置为47.00 nm×47.00 nm×20.00 nm,壁面厚度为1.446 nm,截断半径为1.367 nm,壁面作用势能为1.5 kJ/mol,温度为298 K,正十六烷分子数分别为1 000、1 500、2 000、2 500、3 000。进行模拟计算,可以得到正十六烷纳米液滴的二维密度分布图,根据二维密度图,再采用文献[10]的方法,计算得到的接触角如图8所示。

图8 分子数对接触角的影响
由图8可以看出,对于壁面作用势能为1.5 kJ/mol的光滑壁面,正十六烷分子数在1 000~3 000范围内,模拟得到的接触角均为30°左右,即分子数对接触角影响不大。因此,本文的模拟研究,选用的分子数为2 000个。
2.1.4截断半径的影响
将模拟盒子设置为47.00 nm×47.00 nm×20.00 nm,壁面厚度为1.446 nm,正十六烷分子数为2 000,壁面作用势能为1.5 kJ/mol,温度298 K,截断半径分别为1.171 5、1.366 7、1.562、1.757 3 nm时,进行模拟计算,可以得到正十六烷纳米液滴的二维密度分布图,根据二维密度图,再采用文献[10]的方法,计算得到的接触角如图9所示。

图9 截断半径对接触角的影响
众所周知,当截断半径较小时,模拟结果不够准确,误差比较大;随着截断半径的增加,模拟结果逐渐接近实际情况;当进一步增大截断半径时,模拟结果的变化不显著。增加截断半径所带来的结果使计算时间随着截断半径的增加迅速增长,带来相应的模拟计算成本增加。由图 9可知,对于壁面作用势能为1.5 kJ/mol的光滑壁面,截断半径在1.171 5~1.757 3 nm范围内,模拟得到的接触角均为30°左右,即截断半径对接触角影响不大。因此,本文的模拟研究,选用的截断半径为1.367 nm。
2.2壁面作用势能的影响
选择47.00 nm×47.00 nm×20.00 nm的模拟盒子,壁面厚度为1.446 nm,正十六烷分子数为2 000,截断半径为1.367 nm,在温度为298 K时,探讨壁面作用势能的影响。壁面作用势能分别取0.2、0.4、0.6、0.9、1.2、1.5、1.8、2.1 kJ/mol时,进行模拟计算,可以得到正十六烷纳米液滴的二维密度分布图,根据二维密度图,再采用文献[10]的方法,计算得到的接触角如图10所示。
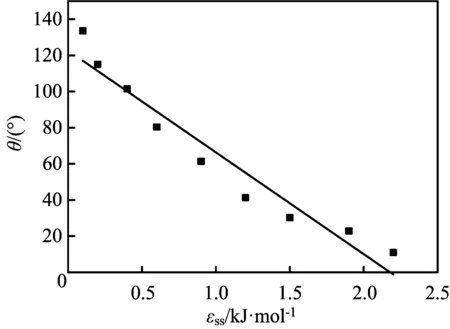
图10 壁面作用势能(εss)对接触角的影响
由图10可见,壁面作用势能的增加,液滴接触角呈线性减小,当壁面作用势能在0.2~2.1 kJ/mol时,接触角变化范围为10°~133°。当壁面作用势能为0.5 kJ/mol时,接触角约为90°,为中性壁面;当壁面作用势能<0.5 kJ/mol时,接触角>90°,为疏油壁面;当壁面作用势能>0.5 kJ/mol时,接触角<90°,为亲油壁面。
2.3温度的影响
选择47.00 nm×47.00 nm×20.00 nm的模拟盒子,壁面厚度为1.446 nm,正十六烷分子数为2 000,截断半径为1.367 nm,探讨温度对接触角的影响。模拟温度分别为298、323、348、373 K下,进行模拟计算,可以得到正十六烷纳米液滴的二维密度分布图,根据二维密度图,再采用文献[10]的方法,计算得到的接触角如图11所示。

图11 温度对接触角的影响
由图 11可知,无论是疏油壁面、中性壁面还是亲油壁面,随着温度的升高,接触角逐渐减小。根据Yong′s方程[12]:
cosθ=(γsv-γsl)/γlv
(4)
式中:γsv、γsl、γsv分别为固—气、固—液和液—气界面间的界面张力,θ为平衡接触角。根据Mimouri等[13]的研究,可以认为,γlv基本为定值;温度的升高,正十六烷液滴的界面张力γsl降低;与γsl项相比,γsv变化特别小;所以,接触角随着温度的提高而逐渐减小。
3 结论
本文采用分子动力学模拟方法,研究了正十六烷纳米液滴在光滑虚拟壁面上的润湿行为。考察了壁面宽(或长)度、厚度、截断半径及正十六烷分子数,壁面作用势能和温度对接触角的影响规律。模拟结果表明,壁面宽(或长)度、厚度、截断半径及十六烷分子数对接触角影响不大。随着壁面作用势能的增大,接触角线性减小;当壁面作用势能为0.5 kJ/mol时,接触角为90°左右。随着温度的提高,无论是亲油、中性还是疏油壁面,接触角都减小。
[1] Abdel Gawad S,Abdel Shafy M.Pollution control of industrial wastewater from soap and oil industries:a case study[J].Water Science and Technology,2002,46(4-5):77-82.
[2] Hassler B.Accidental versus operational oil spills from shipping in the baltic sea:risk governance and management strategies[J].Ambio,2011,40(2):170-178.
[3] 方 龙,陈国定,刘 登.喷射油滴沉积油膜的流动铺展特性研究[J].机械工程学报,2016,52(23):160-167.
[4] Chai J,Liu S,Yang X. Molecular dynamics simulation of wetting on modified amorphous silica surface[J].Applied Surface Science,2009,255(22):9078-9084.
[5] 邱 丰,王 猛,周化光,等.Pb液滴在Ni基底润湿铺展行为的分子动力学模拟[J].物理学报,2013,62(12):1-7.
[6] Heermann D W.Computer-simulation methods[M].Berlin:Springer Berlin Heidelberg,1990.
[7] 陈正隆,徐为人,汤立达.分子模拟的理论与实践[M].北京:化学工业出版社,2007.
[8] 颜 群,王宝和.水及其表面活性剂体系汽—液界面行为的分子动力学模拟[J].河南化工,2015,32(4):17-21.
[9] Rigby M,Smith E,Wakehan W,et al.The forces between molecules clarendon[J].Chem Phys Lett,1986,46(3):1-10.
[10] 王宝和,李 群.接触角的研究现状及其在凝胶干燥中的作用[J].干燥技术与设备,2014,12(1):39-46.
[11] 刘光启,马连湘,刘 杰.化学化工物性数据手册(有机卷)[M].北京:化学工业出版社,2002.
[12] 刘天庆,穆春丰,夏松柏,等.滴状冷凝初始液滴的形成机理[J].化工学报,2007,58(4):821-828.
[13] Langroudi S M M,Ghassemi M,Shahabi A,et al.A molecular dynamics study of effective parameters on nano-droplet surface tension[J].Journal of Molecular Liquids,2011,161(2):85-90.
MolecularDynamicsSimulationofWettingBehaviorofn-HexadecaneNanodropletsonSmoothSurfaces
BAILin,WANGBaohe,YUZhijia
(School of Chemical Engineering , Dalian University of Technology , Dalian 116024 , China)
The wetting behavior ofn-hexadecane nanodroplets on smooth surfaces is investigated by molecular dynamics simulation.Simulation results show that surface,thickness length (or width),cut-off radius and then-hexadecane molecular numbers have little influence on the contact angels.With surface potential energy increasing,the contact angles decrease linearly.When surface potential energy is 0.5 kJ/mol,the contact angel is about 90°.With the increasing of simulation temperature,the contact angle decreases gradually.
TB383,O363
A
1003-3467(2017)11-0025-05
Keywordsmolecular dynamics ; simulation ; contact angel ;n-hexadecane
2017-08-17
国家自然科学基金(51376030)
白 麟(1991-),男,在读硕士,从事油水分离及纳米液滴润湿行为的分子动力学模拟研究,E-mail:sneakerhead_bl@qq.com;联系人:王宝和(1959-),男,副教授,从事不同形貌微纳结构的制备、干燥及分子动力学模拟研究,E-mail:wbaohe@163.com。
