碘普罗胺的合成工艺优化研究
2017-11-28赵长阔马家会王先恒熊麦安
赵长阔,马家会,王先恒,2,熊麦安
(1.遵义医学院 药学院药物化学教研室,贵州 遵义 563099;2.遵义医学院 药学院临床药学教研室,贵州 遵义 563099)
碘普罗胺的合成工艺优化研究
赵长阔1,马家会1,王先恒1,2,熊麦安1
(1.遵义医学院 药学院药物化学教研室,贵州 遵义 563099;2.遵义医学院 药学院临床药学教研室,贵州 遵义 563099)
目的摸索一条符合工业生产的碘普罗胺的合成工艺。方法以价廉易得的5-硝基异酞酸为原料,通过还原、碘化以及与SOCl2反应得到5-氨基-2,4,6-三碘异酞酰氯5,再与甲氧基乙酰氯发生酰化,最后依次与3-氨基-1,2-丙二醇和3-甲胺基-1,2-丙二醇反应得到碘普罗胺。结果对于合成路线中的关键步骤进行了工艺优化,6步反应总收率最高达24.5%。中间体及目标化合物均通过1H NMR和13C NMR确证了结构。结论该方法具有原料易得、合成步骤短、避免了柱层析纯化操作以及产物总收率较高等优点。
5-硝基异酞酸;间苯双酰氯;工艺优化;碘普罗胺
碘普罗胺(Iopromide),商品名为优维显(Ultravist),是由德国先灵(Schering)公司开发,于1985年首次在德国上市,临床用作X-射线造影剂[1-2]。化学结构如图1所示,包含2,4,6-三碘-5-酰氨基-1,3-间苯二酰胺的母体结构,为中性、非离子型化合物。碘普罗胺具有水溶性大、造影增强效果好、毒性较低、耐受性也较好等优点,广泛用于数字减影血管造影、尿路、肾动脉、心室、胃肠等造影以及CT扫描等[3]。

图1 碘普罗胺(1)的化学结构式
从国家食药监局公开的药品注册信息显示,德国先灵最早于2003年获得批准向我国出口碘普罗胺,至今仍然没有一家国内制药企业获得该仿制药的生产批件。临床用碘普罗胺全部依赖进口,该原料药合成在国内尚处于研发阶段。
有关碘普罗胺的合成,文献主要报道了两种合成思路及路线[4-5]:(1)以间苯双酰氯衍生物为关键中间体,依次与甲氧基乙酰氯、3-氨基-1,2-丙二醇以及3-甲胺基-1,2-丙二醇形成酰胺键得到目标产物[1]。该路线简洁、操作相对简便易行,但制备中容易产生双取代(Bismer)副产物,且不宜分离。(2)以5-硝基-1,3-间苯二甲酸的单酯衍生物为中间体,该路线可避免副产物Bismer的生成,后续合成研究大多围绕第二种路线,但总体步骤太过冗长、且操作繁琐[6-8]。适合工业化碘普罗胺的合成工艺有待进一步开发。
根据现有文献,首先选取了大多数研究小组采用的第二种路线:以5-硝基-1,3-间苯二甲酸的单酯为关键中间体,合成制备化合物2[1],但依照文献,尝试用铁粉或者催化氢化等还原方法对硝基进行还原,但是产物难以纯化(见图2)。因此,重新选取第一种路线(见图3),以间苯双酰氯衍生物5为关键中间体,通过优化工艺,成功合成了碘普罗胺,涉及的中间体及目标化合物均通过1HNMR和13CNMR确证了结构。
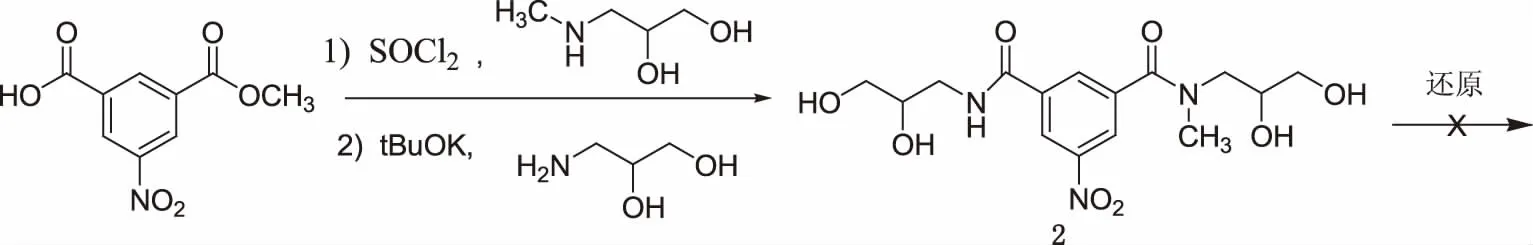
图2 碘普罗胺的合成路线二
1 材料与方法
1.1 材料 5-硝基异酞酸、甲氧基乙酸、3-氨基-1,2-丙二醇、3-甲氨基-1,2-丙二醇、SOCl2(分析纯,国药集团);KI、KIO3、KOH、Na2CO3、冰醋酸、DMF(成都市科龙化工试剂厂);石油醚、乙醚、甲醇、氯仿、二氯甲烷、乙酸乙酯、四氢呋喃、1,4-二氧六环(重庆川东化工集团有限公司);柱层析用硅胶(粒度200~300目)、硅胶薄层板(青岛海洋化工厂)。
仪器采用81-2型暗箱三用紫外分析仪(上海司乐仪器有限公司);旋转蒸发仪(上海箐海仪器有限公司RE-200E型);Varian 400MHz核磁共振波谱仪(TMS作内标,安捷伦科技有限公司);LC-10ATVP 型高效液相色谱仪(日本岛津公司);液相色谱串联质谱联用仪(AB SCIEXL distribution)。
1.2 方法
1.2.1 (N,N’-双(2,3-二羟基丙基)-2,4,6-三碘-5-[(2-甲氧基)乙酰氨基]-N-甲基-1,3-苯二酰胺 (碘普罗胺1)的合成路线 参考文献[1,9],以市场易得的5-硝基异酞酸为原料,通过硝基还原、碘化、以及与SOCl2反应得到间苯双酰氯衍生物 5[9],再与甲氧基乙酰氯发生酰化反应,最后依次与3-氨基-1,2-丙二醇和3-甲胺基-1,2-丙二醇反应得到碘普罗胺1[1];反应路线参考图3。

图3 碘普罗胺的合成路线一
1.2.2 5-氨基异酞酸(化合物3)的制备 取铁粉2.4g 、蒸馏水15.0 mL 置于圆底烧瓶中,快速搅拌下滴加HCl (36%) 0.3 mL,升温回流1 h将铁粉活化后,分批加入5-硝基异酞酸(2.0 g,9.48 mmol) 20 min内加完),加完后100 ℃回流2 h,TLC薄层板跟踪原料消失。反应液冷却至室温,用40%的NaOH (2.0 mL)调节pH至12~14,重新加热,趁热抽滤,热水洗涤滤饼3次,摒弃滤饼,滤液用36%的HCl (2.5 mL)调节至pH1~3,析出无色结晶,将固体产物滤出洗涤后烘干,称重1.51 g,产率87.8%。1H NMR (300 MHz,DMSO-d6),δ (ppm):12.86 (s,2H),7.63 (s,1H),7.35 (s,2H),5.76 (br,2H)。
1.2.3 5-氨基-2,4,6-三碘异酞酸(化合物4)的制备 将水5 mL、5-氨基异酞酸 (0.75 g,4.14 mmol)置于圆底烧瓶,搅拌后得到淡黄色微溶混合物,快速搅拌下加入2.7 mL 36.0%的HCl,再加入碘化钾1.66 g (10 mmol),控制油浴温度在36~41℃,将碘酸钾固体1.07 g (5 mmol) 溶解于水中并缓慢滴加至反应瓶中,反应液颜色变成棕褐色,滴加完成后浴温升至80 ℃,继续搅拌反应过夜,TLC薄层板跟踪原料消失。反应液冷却至室温,析出固体产物,抽滤,滤饼用蒸馏水洗涤,烘干,得到化合物4,称重1.53 g,产率66.2%。1H NMR (400 MHz,DMSO-d6),δ(ppm):13.78 (s,2H),5.56 (s,2H)。
1.2.4 5-氨基-2,4,6-三碘异酞酰氯(化合物5)的制备 取5-氨基-2,4,6-三碘异酞酸(17.21 g,30.8 mmol) 、乙酸乙酯100.0 mL置于干燥的圆底烧瓶中,搅拌下缓慢滴加氯化亚砜(15.0 mL),油浴加热搅拌升温至90 ℃回流,1.5 h后反应液变澄清,4 h后TLC跟踪原料消失。停止反应,减压蒸馏去除乙酸乙酯及过量的氯化亚砜。残留液加入乙酸乙酯(15.0 mL)重新溶解后,加入石油醚,析出固体产物。过滤,干燥,得到目标产物16.0 g,为无色粉末,产率87.2%;Rf=0.91(甲醇/二氯甲烷= 10/1,添加0.5%的冰乙酸);1H NMR (400 MHz,DMSO-d6),δ (ppm):6.06 (s,2H)。
1.2.5 5-甲氧基乙酰氨基-2,4,6-三碘异酞酰氯(化合物6)的制备 取甲氧基乙酸8.9g于干净干燥的反应烧瓶中,加入N,N-二甲基甲酰胺 29.0 mL,冰浴中搅拌。滴加8.4 mL氯化亚砜,然后加入5-氨基-2,4,6-三碘异酞酰氯(10.0 g,16.8 mmol),升温至25~30 ℃反应15 h,将反应液倒入700 mL水中搅拌,固体析出,过滤,50 ℃真空恒温干燥,得到化合物6粗产品10.50 g,将此粗品溶于二氯甲烷中,依次用饱和Na2CO3、5%的氢氧化钠水溶液洗涤,二氯甲烷层用无水Na2SO4干燥后,减压蒸除溶剂后,得到白色固体,真空60 ℃干燥后得到化合物6的纯品9.98 g,产率89.0%。Rf= 0.55 (石油醚:乙酸乙酯=5:3);1H NMR (400 MHz,DMSO-d6),δ (ppm):5.50-6.50 (br,1 H),3.91 (s,2 H),3.33 (s,3 H)。
1.2.6 3-[(2-甲氧基)乙酰氨基]-5-(2,3-二羟基正丙氨基甲酰基)-2,4,6-三碘苯甲酰氯(化合物7)的制备 将5-甲氧基乙酰氨基-2,4,6-三碘异酞酰氯(0.5 g,0.75 mmol)、1,4-dioxane 20 mL置于干燥的圆底烧瓶中,搅拌成悬浮液,冰浴中搅拌下缓慢滴加3-氨基-1,2-丙二醇 (0.075g,0.84mmol),加热回流10 h。将反应液缓慢加入50.0 mL二氯甲烷中,并快速搅拌,析出白色固体,抽滤,真空干燥后化合物7 (0.462g,85.3%)。Rf= 0.32 (展开剂为二氯甲烷:甲醇=10∶1);1HNMR (400 MHz,DMSO-d6),δ (ppm):3.98-4.03(m,1 H),3.67(s,1 H),3.41(d,J=24 Hz,2 H),3.10(s,1H),1.07(s,1H)。
1.2.7 碘普罗胺(化合物1)的制备 将(3-[(2-甲氧基)乙酰氨基]-5-[2,3-二羟基正丙基甲酰胺基])-2,4,6-三碘苯甲酰氯 (674 mg,0.93 mmol)、DMF 1.4 mL置于圆底烧瓶中,溶解搅拌后,冰浴中缓缓滴加3-甲胺基-1,2-丙二醇 (229 mg,2.18 mmol,溶解于0.6 mL DMF中) 滴加完毕后,继续室温搅拌反应,TLC跟踪反应,展开剂为CH2Cl2/CH3OH = 5/2,1.5h后TLC显示原料点消失,停止反应,将反应液缓慢加入50.0 mL二氯甲烷中,并快速搅拌,析出白色固体,抽滤,得到白色固体产物,粗品通过乙醇重结晶后干燥称重470 mg,产率:63.8%,Rf=0.57(展开剂为CH2Cl2/CH3OH=5/1)。1H NMR (400 MHz,DMSO-d6),δ (ppm):8.39(s,1H),7.24(s,1H),4.72(d,J= 28 Hz,2 H),4.48-4.60 (m,2H),3.65-3.88(m,3 H),3.34(s,3 H),3.14(s,4 H),2.95(s,3H),2.82-2.86(m,2H),2.48(s,1H)。13C NMR (100 MHz,DMSO-d6):δ (ppm):170.70,170.26,156.13,156.05,149.94,148.10,91.91,70.54,70.49,70.39,64.38,64.34,60.96,50.56,42.95,37.63,34.53,19.02.MS:[M+1]+=792,HPLC 98.5%。测定条件:层析柱:Agela Venusil MPC-18 (5 μm,4.6×150 mm);流动相:梯度洗脱,65%CH3CN (0.05%TFA)、35%H2O (0.05%TFA)、100% CH3CN (0.05%TFA)。流速:1.0 mL / min;检测波长:254 nm。
2 结果
对于图3的反应路线的每一步骤,通过制备工艺中各项参数优化,得到最佳实验制备条件。
2.1 化合物5的反应条件优化 对于化合物5的制备,专利文献EP091583581采用SOCl2+喹啉+十二烷在80℃反应,反应液后处理时无法完全除去SOCl2,与甲氧基乙酰氯的反应副产物增多,产物不易纯化。研究尝试使用SOCl2在乙酸乙酯中进行回流反应,旋蒸去除溶剂后,用再结晶办法,先将粗品溶解于乙酸乙酯中,再加入不良溶剂石油醚,慢速析出纯净的白色固体产物,收率可达87%。
2.2 化合物6的反应条件优化 由表1得出化合物6的最佳合成条件是:5-氨基-2,4,6-三碘异酰氯、氯化亚砜、甲氧基乙酸的摩尔比为1∶6.88∶5.8;反应时间为15~23 h,温度为25 ℃,产率最佳可达89%。反应时间通过TLC薄层板示踪得出,原料点完全消失即达反应终点。
表1不同摩尔配比对化合物6收率的影响

序号5-氨基-2,4,6-三碘异酞酰氯(摩尔比)氯化亚砜(摩尔比)甲氧基乙酸(摩尔比)时间(h)产率(%)115.486.892487.2215.486.892380.0318.2510.291080.4418.2510.292467.2516.885.81589.0617.065.882388.8
2.3 化合物7的反应条件优化 从结构上来看,化合物6由于含有两个酰氯基团,与3-氨基-1,2-丙二醇进行反应时,如果条件控制不好,理论上会产生较多的双酰化副产物 (bismer),且由于副产物与产物结构较为类似,且极性均较大,产物难以提纯。对于此步反应,我们重点针对3-氨基-1,2-丙二醇与底物的摩尔比,以及反应溶剂的选择进行了多次工艺改进,结果见表2。
表2化合物7的工艺优化

序号摩尔比(3-氨基-1,2-丙二醇/化合物6)溶剂碱时间(h)产率(%)10.79DMF2425.221.0DMF2460.131.1DMF2465.231.5DMF2464.641.1DMF三丁胺2474.151.11,4-二氧六环三丁胺2474.461.11,4-二氧六环几滴DMF1079.171.11,4-二氧六环1085.3
由表中可知,当3-氨基-1,2-丙二醇与底物的摩尔比为1.1,采用1,4-二氧六环为溶剂,不仅收率最高达85%,双酰化副产物 (bismer)含量较少。同时避免了采用高沸点DMF,便于反应后处理。
2.4 步骤六的反应条件优化 选取DMF作为反应溶剂,由表3可知反应条件都相同时,收率不够稳定,其原因与上一步产物纯度有关,反应时间根据TLC现实原料点消失为准。
表3碘普罗胺的工艺优化

序号化合物7投料量(化合物7,g)3-甲氨基-1,2-丙二醇/化合物7(摩尔比)时间(h)收率(%)110.122.34162.1210.32.34142.8310.672.341.563.8410.542.34441.1511.62.30257.5
文献中[1],对碘普罗胺的最后一步制备中,采用离子树脂Amberlite XAD-4来进行产品的纯化,实际上,经该方法纯化得到的碘普罗胺,虽然文献报道收率有69%,但是纯度较低,约60%左右。同时用树脂纯化也不符合工业化生成需求。因此,我们采用简单的重结晶办法处理后,得到了不仅高纯度的碘普罗胺,同时也符合工业生产的需求。
3 讨论
方法[10],以廉价易得的5-硝基异酞酸为原料,通过硝基还原、碘化、以及与SOCl2反应得到间苯双酰氯衍生物5为关键合成中间体,先后与甲氧基乙酰氯、3-氨基-1,2-丙二醇和3-甲胺基-1,2-丙二醇形成酰胺键;成功合成了碘普罗胺,产品色泽好,纯度高;合成的中间体及目标产物均通过1HNMR和13CNMR确证了结构。
在制备碘普罗胺的六步反应中,通过还原、碘化得到化合物4的反应操作简单,且各步收率均较高。本项研究中,重点针对化合物5至目标产物1的合成进行了实验参数优化,主要考察了反应关键底物的投料比、温度、溶剂以及时间等因素对于产物收率的影响(见表1~3);最终得到优化的合成制备条件。合成路线简洁,整体步骤少,终产品收率较高,总收率最高达24.5%。期望能通过后续的放大研究,推动碘普罗胺国产工业化生产,减少进口,从而降低患者用药成本。
[参考文献]
[1] Speck U,Blaszkiewicz P,Seidelmann D,et al.Novel triiodinatedisophthalic acid diamidesasnonionic X-ray contrast media [P].US:4364921A,1982-12-21.
[2] Chao S H,Kim H S,Lee J Y,et a1.Fixed drug eruption due to iopromide (Ultravist) [J].Annals of Dermatology,2011,23(suppl 1):33-35.
[3] 蔡德山.造影剂药物进展与市场透视[J].中国医药技术经济与管理,2008,2(9):16-21.
[4] 赵文龙.碘普罗胺合成路线图解[J].淮海工学院学报:自然科学版,2013,22(3):50-52.
[5] 陆悠玲.碘普罗胺的专利合成路线浅谈[J].广东化工,2015,42(6):1-3.
[6] Hwang K S,Chung S M,Kim C K,et al.Process for preparation of iopromide[P].US:8420858.2013-04-16.
[7] 王哲,陈仕洪,张婷婷,等.碘普罗胺的合成[J].中国医药工业杂志,2012,43(7):527-529.
[8] 张怀义,刘颖平,许志杰,等.碘普罗胺的新制备方法[P].中国:102964269A.2013-03-13.
[9] 谢敏浩,罗世能,何拥军,等.5-甲氧基乙酰胺基-2,4,6-三碘异酞酰氯的合成[J].中国医药杂志,2001,32(3):134-135.
[收稿2017-07-11;修回2017-09-10]
(编辑:王福军)
技术与方法
Researchonthesynthesisprocessoptimizationofiopromide
ZhaoChangkuo1,MaJiahui1,WangXianheng1,2,XiongMaian1
(1.Department of Medicinal Chemistry,School of Pharmacy,Zunyi Medical University,Zunyi Guizhou 563099,China; 2.Department of Clinical Pharmacy,School of Pharmacy,Zunyi Medical University,Zunyi Guizhou 563099,China)
ObjectiveToexplore an industrial manufacture procedure for the active pharmaceutical ingredient (API) Iopromide.MethodsStarting from 5-nitro-isophthalic acid,the key synthetic intermediate of m-benzoyl dichloride 5 was prepared by reduction,iodination and reaction with SOCl2,which reacted in turn with methoxyacetyl chloride,3-amino-1,2-propanediol and 3-methylamino-1,2-propanediol to give iopromide.ResultsThe API iopromide was obtained in total 24.5% of yield with 6 steps.Intermediates and the target compounds were confirmed by1HNMR and13CNMR.ConclusionThe method has the advantages of easy availability of raw materials,involving short synthesis steps,avoiding column chromatography for purification with high total yield of the final product.
5-nitroisophthalic acid; m-benzoyl dichloride; process optimization; Iopromide
遵义医学院自然科学类招标课题([2013]F-680);遵义医学院博士启动基金资助项目(NO:[2014]F697)。
王先恒,女,博士,副教授,研究方向:药物合成及临床药学,E-mail:wangxianheng01@163.com。
R914
A
1000-2715(2017)05-0564-04
