青少年型亨廷顿病10例临床表型及基因突变分析
2017-11-23郝莹陈园园张瑾马惠姿顾卫红
郝莹 陈园园 张瑾 马惠姿 顾卫红
·神经系统遗传性疾病·
青少年型亨廷顿病10例临床表型及基因突变分析
郝莹 陈园园 张瑾 马惠姿 顾卫红
目的总结青少年型亨廷顿病患者临床表型及IT15基因胞嘧啶⁃腺嘌呤⁃鸟嘌呤(CAG)重复突变特点。方法采用聚合酶链反应结合荧光标记毛细管电泳片段分析方法,对159个亨廷顿病家系272名成员进行IT15基因CAG重复序列检测,并对其中10例青少年期发病患者的临床表现、影像学特征以及临床表型与基因型相关性进行分析。结果经基因检测共发现211例携带异常扩展的IT15基因CAG重复序列,其中10例为青少年型亨廷顿病患者,临床表现各异,主要以不自主运动和认知功能障碍为主;发病年龄平均(12.50±4.55)岁,IT15基因CAG重复序列平均(63.70±14.83)个,Pearson相关分析显示,二者呈负相关关系(r=⁃0.865,P=0.001)。结论青少年型亨廷顿病与成年型亨廷顿病患者临床表现不同,前者主要表现为认知功能障碍;对于无明确家族史、临床表现疑似亨廷顿病的患者,基因检测是明确诊断的依据;亨廷顿病发病年龄与IT15基因CAG重复次数呈负相关,但不能完全解释发病年龄的变异性,尤以青少年型亨廷顿病患者显著,可能存在其他遗传调节因素。
杭廷顿病; 青少年; 表型; 基因; 突变; 系谱
亨廷顿病[HD,在线人类孟德尔遗传数据库(OMIM)编号:143100]是常染色体显性遗传性神经变性病,发病年龄30~50岁,病程15~20年,临床主要表现为舞蹈样动作、进行性认知功能减退和神经精神症状,影像学可见尾状核和大脑皮质萎缩[1⁃4]。致病基因IT15定位于4p16.3,第1外显子内存在高度多态性胞嘧啶⁃腺嘌呤⁃鸟嘌呤(CAG)重复序列,该重复序列异常扩展导致亨廷顿病[5]。亨廷顿病发病率为5~8/10万[6],通常于成年期发病,具有明显的外显不全和延迟外显。青少年型亨廷顿病较为少见,临床表型存在异质性。本研究回顾分析10例青少年型亨廷顿病患者的临床资料,对其临床表型和IT15基因CAG重复突变进行细致分析。
对象与方法
一、研究对象
研究对象均来自2005年8月-2016年5月中日友好医院运动障碍与神经遗传病研究中心收集的159个临床拟诊为亨廷顿病家系共272名成员,均为汉族,男性143名,女性129名;年龄6~72岁,平均(36.76±13.73)岁。本研究经中日友好医院道德伦理委员会审核批准,所有受试者及其家属均知情同意并签署知情同意书。
二、研究方法
1.样本采集 空腹采集所有受试者外周静脉血5 ml,以质量分数为3.8%枸橼酸钠进行抗凝,标准酚氯仿法提取基因组DNA。
2.聚合酶链反应 根据IT15基因第1外显子CAG重复片段两侧的序列设计引物,由北京赛百盛基因技术有限公司合成,正向引物(F引物)序列:5'⁃AGTAAGGCCTTCGAGTCCCTCAAGTCCTCC ⁃3';反向 引 物(R引 物)序 列 :5'⁃AAACTCACGGTCGGTGCAGCGGCTCCTCAG ⁃3'。PCR 反应体系共 25 μl,依次加入 dNTPs 2.50 mmol、2×GC缓冲液Ⅰ12.50 μl、正向引物和反向引物分别5 pmol、模板 DNA 100 ng和r⁃Taq 1U,加灭菌去离子水补充至25 μl;反应条件为95℃预变性5 min,95 ℃ 90 s、62.5 ℃ 60 s、72 ℃ 120 s,共循环35次,72℃延伸10 min。PCR扩增产物行质量分数为1.5%的琼脂糖凝胶电泳共40 min,电压100 V,对出现2条电泳条带的样本进行基于毛细管电泳的片段分析。
3.基于毛细管电泳的片段分析 采用荧光标记引物进行PCR扩增反应,引物序列包括经D4荧光标记的上述正向引物和反向引物,其中D4荧光标记的正向引物由生工生物工程(上海)股份有限公司合成。PCR反应体系共计25 μl,依次加入dNTPs 2.50 mmol、2× GC 缓冲液Ⅰ12.50 μl、正向引物和反向引物各5 pmol、模板 DNA 100 ng和r⁃Taq 1U,加灭菌去离子水补充至25 μ l。采用美国Beckman Coulter公司生产的CEQ8000核酸分析仪对PCR扩增产物进行片段分析:甲酰胺 20 μl,以 0.25 μl CEQ DNA Size Standard Kit⁃600片段作为标准内标,异源双链聚合酶链反应(HD⁃PCR)扩增产物1 μl混匀、上样;预设程序进行电泳分离,分离条件为毛细管温度达50℃时,90℃变性120 s,2 kV电压下注入样本30 s、4.80 kV电压下电泳70 min,以预设的分析参数进行片段分析。
4.统计分析方法 采用SPSS 13.0统计软件进行数据处理与分析。采用Kolmogorov⁃Smirnov检验和Shapiro⁃Wilk检验(N<50)行正态性检验;呈正态分布的计量资料以均数±标准差(x±s)表示,发病年龄与IT15基因CAG重复序列的相关分析采用Pearson相关分析。以P≤0.05为差异具有统计学意义。
结 果
基因检测显示,159个亨廷顿病家系共272名成员中211例携带异常扩展的IT15基因CAG重复序列,其中已发病且临床资料完整患者149例,男性83例,女性66例;年龄9~69岁,平均(42.52±12.81)岁;发病年龄 6~66岁,平均为(38.25±12.34)岁;IT15基因CAG重复序列36~80个,平均(45.10±7.55)个。Pearson相关分析显示,发病年龄与IT15基因CAG重复序列呈负相关(r=⁃0.750,P=0.000)。211例携带异常扩展CAG重复序列的患者中10例于青少年期发病,9例为父系遗传,其中7例有明确家族史,余1例为母系遗传。10例青少年型亨廷顿病患者的临床资料参见表1。
一、临床特征
例1(患者3) 男性,19岁,主因不自主运动伴震颤2~3年,于2008年5月12日至我院运动障碍与神经遗传病研究中心就诊。患者3年前无明显诱因出现不自主运动伴震颤,行走易跌倒,易受惊吓。门诊神经系统检查:神志清楚,语言流利,反应尚可,面部有摔伤;眼球各向活动充分,未见明显眼震;四肢肌力5级、肌张力正常,双侧指鼻试验明显意向性震颤,双侧跟⁃膝⁃胫试验尚稳准,直线行走稍差,腱反射正常,病理征阴性。影像学检查:头部MRI显示,大脑皮质轻度萎缩,尾状核头部明显萎缩(图1a)。患者家系可追溯4代共2例患者,均为男性,呈常染色体显性遗传。其父现年41岁,于35岁发病,神经系统检查可见不自主运动,双侧指鼻试验稳准、双侧快复轮替动作笨拙、直线行走稍差。
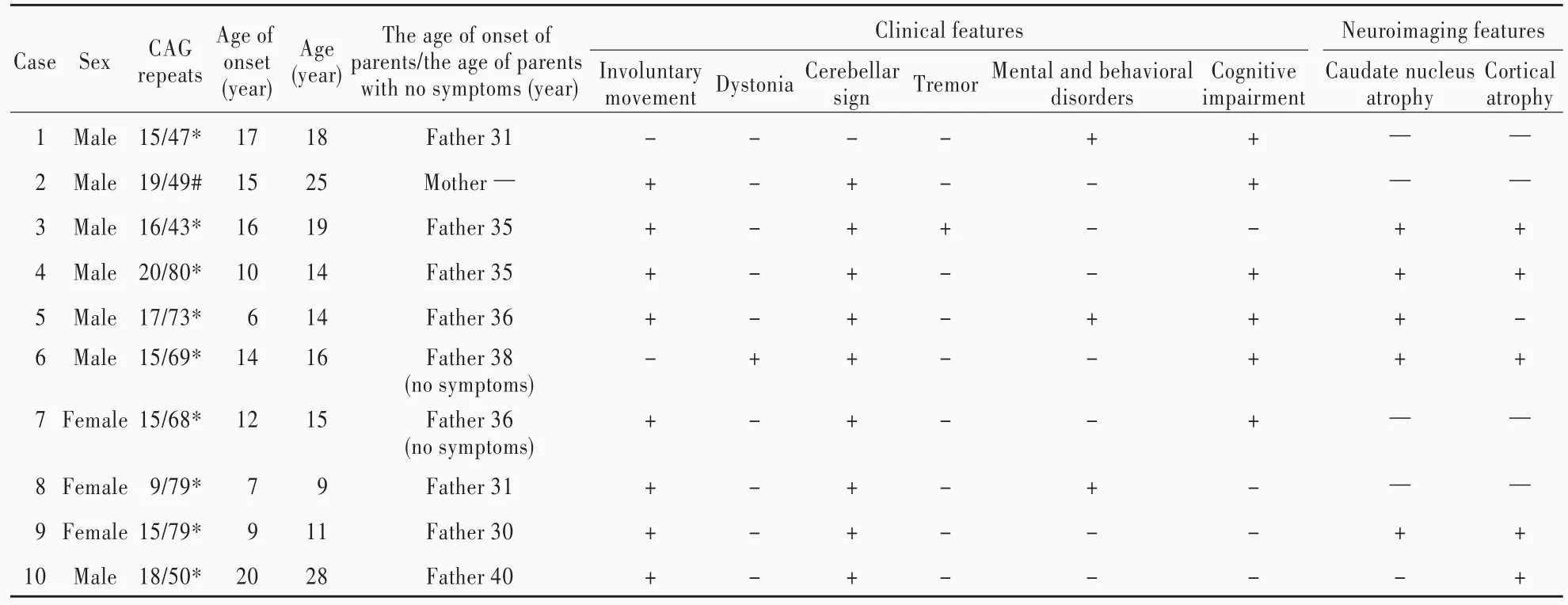
表1 10例青少年型亨廷顿病患者临床资料Table 1. The clinical features of 10 cases with juvenile⁃onset HD
例2(患儿4) 男性,14岁,主因间断性不自主耸肩、动作不协调4年,于2008年7月23日至我院运动障碍与神经遗传病研究中心就诊。患儿4年前无明显诱因出现不自主耸肩,行走不协调,向左侧倾倒,双腿僵硬,睡眠欠佳,睡眠中踢腿、打拳等动作较多。门诊神经系统检查:神志清楚,言语欠流利,智力下降;眼球各向活动缓慢,未见明显眼震,可见角膜色素环;四肢肌力5级,上肢肌张力5级、下肢肌张力增高,双侧指鼻试验和跟⁃膝⁃胫试验欠稳准,腱反射活跃,病理反射未引出。影像学检查:头部MRI显示,大脑皮质中度萎缩,尾状核头部明显萎缩(图1b)。患者家系可追溯3代共4例患者,男性3例,女性1例,呈常染色体显性遗传。其父于35岁发病,表现为不自主运动、行走不稳,于43岁因心肌梗死死亡;其姑母亦表现出相似症状。
例3(患儿6) 男性,16岁,主因运动功能下降伴智力减退2年,于2009年9月8日至我院运动障碍与神经遗传病研究中心就诊。患儿2年前无明显诱因出现动作笨拙、运动功能下降、智力减退等症状与体征,进行性加重。神经系统检查:神志清楚,言语欠流利,反应迟钝;双眼上下视不能,眼球各向活动缓慢,未见明显眼震;四肢肌力5级、肌张力增高,双侧指鼻试验笨拙但尚稳准,双侧快复轮替动作笨拙,行走时可见肌张力障碍姿势,颈部和肩部歪斜,腱反射亢进,病理反射未引出。影像学检查:头部MRI显示,大脑皮质轻度萎缩,尾状核头部轻度萎缩(图1c)。无明确家族史,其父现年38岁,其母40岁,均无相似临床症状,神经系统检查未见明显异常。
例4(患者10) 男性,28岁,主因行走不稳伴言语模糊8年,于2015年5月27日至我院运动障碍与神经遗传病研究中心就诊。患者8年前无明确诱因出现行走不稳、言语模糊,反应较慢,不自主抖动,进行性加重。门诊神经系统检查:神志清楚,口吃,构音障碍;面部不自主运动,姿势异常,四肢小幅度不自主抖动;眼球各向活动欠充分,未见明显眼震;四肢肌力5级、肌张力增高,双侧快复轮替动作笨拙,双侧指鼻试验和跟⁃膝⁃胫试验欠稳准,腱反射对称,病理征可疑阳性。影像学检查:头部MRI显示,大脑皮质轻度萎缩,尾状核头部中度萎缩(图1d)。患者家系可追溯4代共4例患者,男性2例,女性2例,呈常染色体显性遗传。其父现年53岁,于40岁发病,就诊时全身呈大幅度不自主运动;其他家系成员无法追溯。

图1 4例青少年型亨廷顿病患者头部MRI检查所见 1a 例1(患者3)横断面T1WI显示大脑皮质轻度萎缩(粗箭头所示),尾状核头部明显萎缩(细箭头所示) 1b 例2(患儿4)横断面T2WI显示大脑皮质中度萎缩(粗箭头所示),尾状核头部明显萎缩(细箭头所示) 1c 例3(患儿6)横断面T2WI显示大脑皮质轻度萎缩(粗箭头所示),尾状核头部轻度萎缩(细箭头所示) 1d 例4(患者10)横断面T2WI显示大脑皮质轻度萎缩(粗箭头所示),尾状核头部中度萎缩(细箭头所示)Figure 1 Head MRI findings of 4 patients with juvenile⁃onset HD Axial T1WI of Case 3 showed mild atrophy of cerebral cortex(thick arrow indicates)and obvious atrophy of the head of caudate nucleus(thin arrow indicates,Panel 1a).Axial T2WI of Case 4 showed moderate atrophy of cerebral cortex(thick arrow indicates)and obvious atrophy of the head of caudate nucleus(thin arrow indicates,Panel 1b).Axial T2WI of Case 6 showed mild atrophy of cerebral cortex(thick arrow indicates)and the head of caudate nucleus(thin arrow indicates,Panel 1c).Axial T2WI of Case 10 showed mild atrophy of cerebral cortex(thick arrow indicates)and moderate atrophy of the head of caudate nucleus(thin arrow indicates,Panel 1d).
二、IT15基因CAG重复片段分析
共10例青少年型亨廷顿病患者,男性7例,女性3例;年龄9~28岁,平均(32.00±5.88)岁;发病年龄 6~20岁,平均(12.50±4.55)岁;IT15基因CAG重复序列43~80个,平均(63.70±14.83)个。Pearson相关分析显示,发病年龄与IT15基因CAG重复序列呈负相关(r=⁃0.865,P=0.001)。
讨 论
亨廷顿病是一种以运动障碍为突出表现的常染色体显性遗传性神经变性病,多于中年期隐匿起病,临床表现除进行性加重的舞蹈样动作、认知功能障碍和行为异常“三联征”外,还伴有共济失调、吞咽困难和构音障碍[7],主要病变部位位于纹状体(包括尾状核和壳核)和大脑皮质,病残率较高,严重影响患者生活质量,目前全世界范围内其发病率为5~10/10万[8]。亨廷顿病最早于1872年由Huntington[9]系统描述而得名,于 1993年克隆出其致病基因IT15基因,定位于4p16.3,包含67个外显子,其mRNA包含13 474个核苷酸,编码1个包含3142 个氨基酸残基的蛋白即亨廷顿蛋白(Htt)[10⁃11],其第1外显子内存在一段高度多态性CAG重复序列,该重复序列异常扩展可以导致亨廷顿病。
亨廷顿病发病年龄为24~46岁,平均(35.8±11.8)岁;病程 7~ 16年,平均(11.6± 5.6)年[12],约25%患者于50岁甚至70岁后发病,临床症状轻微。成年型亨廷顿病临床表现多以运动障碍、智力减退和精神行为异常为主;青少年型亨廷顿病临床少见,发病率0.50 ~ 1.00/10万,占成年型的1/10[13],多为父系遗传,20岁前发病,临床表现与成年型不同,主要表现为运动障碍、肌强直、腱反射亢进、眼动异常、严重认知功能障碍,特别是10岁前发病患者,临床常出现帕金森样表现,如肌张力障碍、震颤、运动迟缓等。本研究经基因检测确定159个亨廷顿病家系共211例患者携带异常扩展的CAG重复序列,其中149例已发病,绝大多数于成年期发病,仅10例于青少年期发病,8例表现为小脑体征、7例出现不自主运动、6例出现认知功能障碍、3例出现精神行为异常、1例出现震颤、1例无明确家族史患者出现肌张力障碍。10例患者中4例发病年龄≤10岁,分别为10、6、7和9岁;IT15基因CAG重复序列分别为80、73、79和79次,其中,10岁发病的患儿4临床表现为不自主运动、行走不稳、智力下降、睡眠中出现打拳、踢腿等动作;6岁发病的患儿5表现为行走姿势异常、足内翻、双上肢和面部不自主抖动、智力下降、反应较慢、情绪异常;7岁发病的患儿8表现为行走易跌倒、不自主运动、学习成绩较差、性格孤僻;9岁发病的患儿9表现为双侧快复轮替动作笨拙、不自主运动,上述4例患儿均无舞蹈样动作。国外曾报道多例青少年型亨廷顿病患者,例如,Yoon等[14]报告3例10岁前发病的患儿,CAG重复序列分别为120、100和93次,早期表现为精神行为异常、烦躁、多动、共济失调、易跌倒,后期均出现严重构音障碍。Ribai等[15]研究显示,29例法国青少年型亨廷顿病患者中19例(65.52%)最常见症状是严重认知功能障碍和精神行为异常,包括酒精和药物依赖及精神障碍,平均发生于发病后6年;3例出现肌阵挛伴头部震颤,3例为舞蹈样动作,1例为进行性小脑共济失调;10岁前发病的患者均为父系遗传。Patra和 Shirolkar[16]报告 1 例 IT15 基因 CAG重复序列为83次的青少年型亨廷顿病患者,8岁发病,临床表现为严重癫发作、共济失调、运动迟缓、易跌倒,无明确家族史,病情进展较快,9岁时出现严重抽搐发作,病程中无舞蹈样动作;头部MRI显示双侧尾状核对称性T2WI高信号,轻度萎缩。我们研究团队曾报告1例少年型亨廷顿病患儿,其父未发病,但是携带CAG中间重复等位基因[17]。青少年型亨廷顿病与成年型亨廷顿病患者临床表型的差异提示受累神经核团不完全相同,可能与神经系统发育过程有关。
临床上有些疾病表型与亨廷顿病极为相似,应注意鉴别诊断。(1)类亨廷顿病1型(HDL1型):呈常染色体显性遗传,发病年龄21~34岁,致病基因为PRNP基因;临床以精神症状为主,此外还表现为共济失调、构音障碍、协调能力差、认知功能障碍、痴呆、舞蹈样动作、面部不自主运动和肌强直等[18]。(2)类亨廷顿病2型(HDL2型):呈常染色体显性遗传,发病年龄35~40岁,致病基因为JPH3基因,其变位剪切体处存在一段多态性CAG重复序列,正常重复次数为6~28次,≥41次为异常重复次数;临床主要表现为肌张力障碍、舞蹈样动作、肌强直、构音障碍、运动迟缓、痴呆、动作性震颤和精神症状等;头部影像学显示纹状体萎缩[19]。(3)类亨廷顿病3型(HDL3型):呈常染色体隐性遗传,发病年龄为3~5岁,致病基因定位于4p15.3;临床表现为共济失调、痉挛、舞蹈样动作、肌张力障碍、癫发作、锥体外系症状和精神症状等;头部影像学显示尾状核和额叶皮质萎缩[20]。(4)类亨廷顿病4型(HDL4型):又称脊髓小脑共济失调17型(SCA17型),呈常染色体显性遗传,发病年龄19~48岁,致病基因为TBP基因,其第3外显子区存在一段多态性CAG重复序列,正常重复次数为25~42次,异常重复次数为45~66次;临床表现为共济失调、构音障碍、意向性震颤、精神症状、痴呆、肌阵挛、肌张力障碍和舞蹈样动作等[21]。
多项研究显示,CAG重复序列<27次为正常等位基因,不引起疾病;27~35次为可引起突变的等位基因,不引起疾病,但在减数分裂过程中易发生扩展突变,使后代致病;36~40次易发生外显不全,其根本原因是代间传递不稳定,使后代重复发生明显扩展,携带者可能发病也可能不发病;>40次为完全外显的异常等位基因,引起疾病[22⁃26]。对于无明确家族史的散发病例,应通过基因检测明确诊断,同时还应对其父母进行基因检测,以免漏诊。
发病年龄与扩展的CAG重复序列呈负相关,但仅能解释50%~70%的发病年龄变异性。有文献报道,青少年型亨廷顿病患者CAG重复序列>50次,成年期发病患者CAG重复序列约为40次[27]。本研究家系中211例携带异常扩展的IT15基因CAG重复序列的患者中149例已发病,CAG重复序列约为40次的患者多于成年期发病,如携带43次CAG重复序列的患者发病年龄多≥40岁。本研究10例青少年型亨廷顿病患者中4例CAG重复序列≤50次,分别为47、49、43和50次,相对应的发病年龄分别为17、15、16和20岁,尤其是重复43次的患者16岁即发病,提示青少年型亨廷顿病患者存在其他调节因素影响发病年龄,有学者认为可能与DNA稳定性相关遗传调节因素有关[28]。
亨廷顿病是一种常染色体显性遗传性神经变性病,逐渐出现运动障碍、认知功能障碍和情感障碍。由于越来越多的家系后代逐渐具备一定的医学常识,看到上一代病情后,难免产生恐惧心理。目前该病尚无有效治疗方法,基于对家系患者的明确诊断开展产前诊断和植入前诊断,中断致病突变的传递,对家系后代十分重要。此外,康复训练和心理指导对神经变性病患者及其家属也是至关重要的,亨廷顿病患者应尽早开展运动功能和认知功能康复,对于情感和精神障碍,应尽早予心理指导;家属也应充分了解该病特点和病程规律,采取相应的照料,亦需要心理支持。国外最新文献报道,神经变性病动物模型显示,海藻糖具有抑制异常蛋白质聚集、保护神经细胞的作用,可能对于多聚谷氨酰胺类疾病具有一定延缓疾病发展的作用[29]。与此同时,深入研究亨廷顿病相关遗传调节因素,对发病机制和干预治疗的探索具有重要意义。
[1]Novak MJ,Tabrizi SJ.Huntington's disease.BMJ,2010,340:C3109.
[2]Mason S,Barker RA.Rating apathy in Huntington's disease:patients and companions agree.J Huntingtons Dis,2015,4:49⁃59.
[3]Wood NI,Morton AJ.Social behaviour is impaired in the R6/2 mouse model of Huntington's disease.J Huntingtons Dis,2015,4:61⁃73.
[4]Roos RA.Huntington's disease:a clinical review.Orphanet J Rare Dis,2010,5:40.
[5]Yan YP,Zhang BR.The molecular mechanisms of Huntington's disease and its advances in therapeutic strategies.Zhongguo Xian Dai Shen Jing Ji Bing Za Zhi,2011,11:30⁃35[.严雅萍,张宝荣.亨廷顿病的发病机制和治疗进展.中国现代神经疾病杂志,2011,11:30⁃35.]
[6]Pringsheim T,Wiltshire K,Day L,Dykeman J,Steeves T,Jette N.The incidence and prevalence of Huntington's disease:a systematic review and meta⁃analysis.Mov Disord,2012,27:1083⁃1091.
[7]Smith MA, Brandt J, Shadmehr R. Motor disorder in Huntington's disease begins as a dysfunction in error feedback control.Nature,2000,403:544⁃549.
[8]Gonzalez⁃Alegre P,AfifiAK.Clinicalcharacteristics of childhood⁃onset(Juvenile)Huntington disease:report of 12 patients and review of the literature.J Child Neurol,2006,21:223⁃229.
[9]Huntington G.On chorea.Med Surg Rep,1872,26:320⁃321.
[10]JohriA,BealMF.Antioxidants in Huntington's disease.Biochim Biophys Acta,2012,1822:664⁃674.
[11]Pla P,Orvoen S,Saudou F,David DJ,Humbert S.Mood disorders in Huntington's disease:from behavior to cellular and molecular mechanisms.Front Behav Neurosci,2014,8:135.
[12]Zheng Z,Burgunder JM,Shang H,Guo X.Huntington's like conditions in China,a review of published Chinesecases.Version 2.PLoS Curr,2012,4:RRN1302.
[13]Nance MA,Myers RH.Juvenile onset Huntington's disease⁃clinical and research perspectives.Ment Retard Dev Disabil Res Rev,2001,7:153⁃157.
[14]Yoon G,Kramer J,Zanko A,Guzijan M,Lin S,Foster⁃Barber A, Boxer AL. Speech and language delay are early manifestations of juvenile⁃onset Huntington disease.Neurology,2006,67:1265⁃1267.
[15]Ribai P,Nguyen K,Hahn⁃Barma V,Gourfinkel⁃An I,Vidailhet M,LegoutA,Dodé C,BriceA,DürrA.Psychiatricand cognitive difficulties as indicators of juvenile huntington disease onset in 29 patients.Arch Neurol,2007,64:813⁃819.
[16]Patra KC, Shirolkar MS. Childhood⁃onset (Juvenile)Huntington's disease:a rare case report.J Pediatr Neurosci,2015,10:276⁃279.
[17]Hao Y,Chen YY,Gu WH,Wang GX,Ma HZ,Li LL,Wang K,Jin M,Duan XH.Clinical and genetic study of a juvenile⁃onset Huntington disease.Zhongguo Xian Dai Shen Jing Ji Bing Za Zhi,2012,12:288⁃293[.郝莹,陈园园,顾卫红,王国相,马惠姿,李丽林,王康,金淼,段晓慧.少年型亨廷顿病临床与基因突变分析.中国现代神经疾病杂志,2012,12:288⁃293.]
[18]Laplanche JL,Hachimi KH,Durieux I,Thuillet P,Defebvre L,Delasnerie ⁃LauprêtreN,Peoc'h K,Foncin JF,DestéeA.Prominent psychiatric features and early onset in an inherited prion disease with a new insertional mutation in the prion protein gene.Brain,1999,122(Pt 12):2375⁃2386.
[19]Holmes SE,O'Hearn E,Rosenblatt A,Callahan C,Hwang HS,Ingersoll⁃Ashworth RG,Fleisher A,Stevanin G,Brice A,Potter NT,Ross CA,Margolis RL.A repeat expansion in the gene encoding junctophilin⁃3 is associated with Huntington disease⁃like 2.Nat Genet,2001,29:377⁃378.
[20]Kambouris M,Bohlega S,Al⁃Tahan A,Meyer BF.Localization of the gene for a novel autosomal recessive neurodegenerative Huntington⁃like disorder to 4p15.3.Am J Hum Genet,2000,66:445⁃452.
[21]Zhang J,Hao Y,Gu WH,Chen YY,Wang GX,Wang K,Jin M,Duan XH.Molecularand clinicalstudy ofspinocerebellar ataxia type 17.Zhonghua Shen Jing Ke Za Zhi,2012,45:861⁃865[.张瑾,郝莹,顾卫红,陈园园,王国相,王康,金淼,段晓慧.脊髓小脑共济失调17型临床特征和基因突变分析.中华神经科杂志,2012,45:861⁃865.]
[22]ACMG/ASHG Statement,theAmerican CollegeofMedical Genetics/American Society ofHuman Genetics Huntington Disease Genetic Testing Working Group.Laboratory guidelines for Huntington disease genetic testing.Am J Hum Genet,1998,62:1243⁃1247.
[23]Walker FO.Huntington's disease.Lancet,2007,369:218⁃228.
[24]Losekoot M,van Belzen MJ,Seneca S,Bauer P,Stenhouse SA,Barton DE;European MolecularGenetic Quality Network(EMQN). EMQN/CMGS best practice guidelines for the molecular genetic testing of Huntington disease.Eur J Hum Genet,2013,21:480⁃486.
[25]Wang YN,Li J,Wu J,Bao Y,Yu ZR,Nan SJ.A case report of Huntington's disease.Zhongguo Shi Yan Zhen Duan Xue,2016,20:130⁃131[.王一楠,李君,吴杰,鲍跃,于滋润,南善姬.亨廷顿舞蹈病一家系报道.中国实验诊断学,2016,20:130⁃131.]
[26]Jin FY,Zhang BR.Progress in studies of gene therapy for Huntington's disease.Zhongguo Xian Dai Shen Jing Ji Bing Za Zhi,2012,12:238⁃244[.金范莹,张宝荣.亨廷顿病基因治疗研究进展.中国现代神经疾病杂志,2012,12:238⁃244.]
[27]Rao R,Han YS,Xue BC,Xu Y,Cheng N,Han YZ.Clinical analysis of 11 cases with genetic diagnosis of Huntington's disease.Zhongguo Lin Chuang Shen Jing Ke Xue,2014,22:65⁃69[.饶娆,韩永升,薛本春,徐银,程楠,韩永竹.经基因诊断证实的11例亨廷顿舞蹈病患者临床特点分析.中国临床神经科学,2014,22:65⁃69.]
[28]Yang CR,Yu RK.Intracerebral transplantation of neural stem cells combined with trehalose ingestion alleviates pathology in a mouse model of Huntington's disease.J Neurosci Res,2009,87:26⁃33.
[29]Tanaka M,Machida Y,Nukina N.A novel therapeutic strategy for polyglutamine diseasesby stabilizing aggregation⁃prone proteins with small molecules.J Mol Med(Berl),2005,83:343⁃352.
Clinical and genetic analysis of juvenile⁃onset Huntington's disease:10 cases report
HAO Ying1,CHEN Yuan⁃yuan1,ZHANG Jin1,MA Hui⁃zi2,GU Wei⁃hong11Movement Disorder&Neurogenetics Research Center,China⁃Japan Friendship Hospital,Beijing 100029,China
2Department of Neurology,Beijing Tiantan Hospital,Capital Medical University,Beijing 100050,China
Corresponding authors:MA Hui⁃zi(Email:mahuizi1127@126.com);
GU Wei⁃hong(Email:jane55.gu@vip.sina.com)
ObjectiveTo investigate the clinical features and dynamic mutation of 10 cases with juvenile⁃onset Huntington's disease(HD).MethodsThe cytosine⁃adenine⁃guanine(CAG)repeats of IT15 gene were detected by polymerase chain reaction(PCR)and capillary electrophoresis in 272 individuals of 159 pedigrees with preliminary diagnosis of HD.The correlation between clinical features and expanded CAG repeats in the IT15 gene of 10 cases with juvenile⁃onset HD were studied carefully.Results Among 211 individuals carried expanded CAG repeats,10 cases onset before 20 years of age.The predominant clinical manifestations were involuntary movement and cognitive impairment.The average age of onset was(12.50±4.55)years,and the average CAG repeat number of IT15 gene was 63.70±14.83.Pearson correlation analysis showed that the age of onset was significantly and negatively correlated with the CAG repeat number(r= ⁃0.865,P=0.001).Conclusions1)The juvenile⁃onset case of HD presented with different clinical features compared with adult⁃onset cases.The most common presentation is cognitive decline.2)Analysis of CAG repeats of IT15 gene is necessary for the diagnosis of juvenile⁃onset case of HD with no family history.3)The variability in age of onset is not completely explained by the effects of expanded CAG repeats of IT15 gene,which is more prominently for the juvenile⁃onset cases,therefore,it is suggested that other factors may modulate the age of onset.
Huntington disease; Adolescent; Phenotype; Genes; Mutation; Pedigree
This study was supported by Grant Awarded 2010-2012 from Ministry of Health Foundation of China.
10.3969/j.issn.1672⁃6731.2017.08.008
卫生部部署(管)医院2010-2012年度临床学科重点项目
100029北京,中日友好医院运动障碍与神经遗传病研究中心(郝莹,陈园园,张瑾,顾卫红);100050首都医科大学附属北京天坛医院神经内科(马惠姿)
马惠姿(Email:mahuizi1127@126.com);顾卫红(Email:jane55.gu@vip.sina.com)
2017⁃06⁃14)
