美国药品上市后研究的监管制度及其对我国的启示Δ
2017-11-16罗雪燕赖寒陈绍成李俊重庆第二师范学院生物与化学工程系重庆400067
罗雪燕,赖寒,陈绍成,李俊(重庆第二师范学院生物与化学工程系,重庆400067)
美国药品上市后研究的监管制度及其对我国的启示Δ
罗雪燕*,赖寒,陈绍成,李俊#(重庆第二师范学院生物与化学工程系,重庆400067)
目的:为完善我国Ⅳ期临床试验的监管提出建议。方法:通过概述美国药品上市后研究制度,分析美国FDA对药品上市后研究的监管(包括关键要素、监管流程、配套监管系统和强制措施),提出完善我国Ⅳ期临床试验监管的建议。结果与结论:美国药品上市后研究包括上市后承诺研究(PMR)和上市后要求研究(PMC)。监管过程中的关键要素包括监管主体(由药品评价和研究中心下属的新药办公室负责)、关键文件(包括帮助FDA和申请人达成研究协议的文件和用于对已确定的研究进行过程跟踪和监督的文件)、重要时间节点(明确提交相关材料的特定节点日期);监管流程包括制订研究草案、审核研究报告;美国FDA建立了PMC/PMR数据库作为配套监管系统,并分别针对PMC和PMR制定了相应的强制措施。我国相关监管部门应转换监管思路、充分发挥政府的引导和监督作用,加强药品上市前、后监管的衔接,制订特色化Ⅳ期临床方案,建立Ⅳ期临床试验数据管理系统,加强全过程监管,借鉴FDA“事前制定计划,事中动态追踪,事后依法处理”的监管方式来完善对我国Ⅳ期临床试验的监管。
上市后承诺研究;上市后要求研究;监管;Ⅳ期临床试验;美国
药品上市前虽然经历了严格的药学、非临床和临床研究,但药品上市的决定只是基于已有的非临床和临床数据进行风险评估之后的平衡决策结果,作为决策基础的试验数据也有其固有缺陷[1-2]。因此,与我国一样,美国FDA在批准新药上市的同时,或者在批准药品上市后,只要认为有必要获得更多有关药品风险、利益及最佳使用方法等信息资料,就可以依照法规授权,建议或要求申请人进行药品上市后研究[3]。而对于这类“研究”,美国《食品药品管理补充法案》(2007年)将其区分为“临床试验(Clinical trials)”和“研究(Study)”两类[4],并最终将其统一界定为药品的“上市后研究和临床试验”(Post-marketing clinical trial and study,以下简称“上市后研究”)制度[5]。
笔者拟通过文献研究,从药品监管者的角度出发,围绕监管中的关键要素、监管流程、配套监管系统和强制措施四个方面,系统分析美国FDA对药品上市后研究的监管方式。在此基础上,借鉴美国FDA的监管经验,对完善我国Ⅳ期临床试验监管制度提出建议。
1 美国药品上市后研究制度概述
美国药品上市后研究制度是指针对上市后药品的安全性和有效性研究,包括临床试验、调查、动物研究以及实验室研究等。具体来说,即根据医药学最新学术水平,从药理学、药剂学、临床医学、药物流行病学及药物政策等方面,对已批准的药品在社会人群中的疗效、不良反应、用药方案、稳定性、费用等多方面是否符合安全、有效、经济合理等原则作出科学的评价[6]。根据研究是否具有法律强制性,药品上市后研究可分为上市后承诺研究(Post-marketing commitment research,PMC)和上市后要求研究(Post-marketing requirement research,PMR)。
1.1 PMC
根据美国国会1997年通过的《1997年食品药品管理现代化法》(Food and Drug Administration Moderization Act of 1997,FDCA)”,以及2002年通过的《2002年公共健康安全和生物恐怖预案应对法》中对FDCA 506B条款有关药品上市后研究的补充,规定药品PMC,即药品上市后需要开展的非法定的、未经FDA强制要求的相关研究属协议任务。研究内容一般由FDA提出,需要上市药品责任人同意并承诺进行研究,该行为不受法律约束,申报者仅需每年向FDA报告研究的实施进程[7]。
PMC的目的在于对潜在风险较大的药品的风险、效益比进行持续、动态评估,以更好地保障患者用药安全。其研究内容相对宽泛,多是为了获得更多药品风险、效益及最佳使用信息,解决上市使用中出现的安全性问题和顾虑,包括对药物的疾病史与不良反应发生率的流行病学研究,进一步确定疗效的临床研究和进一步的质量研究等[8]。
1.2 PMR
2007年,美国签署并发布了《食品药品监督管理局2007修正法案》(Food and Drug Administration Amendments Act of 2007,FDAAA)[9],其第9部分“加强对药品上市后的安全监管”中的505(o)条授权FDA,允许FDA在处方新药或新生物制品上市批准的同时,或在批准上市后,当意识到药品的新安全性信息或对药品安全性存在威胁时,可以依法要求上市药品责任人进行该药物或生物制品的上市后研究。
上述条款赋予了FDA更多用于药品上市后安全监管和风险管理的权力和资金,标志着FDA的这一权力和职能正式获得法律认可。然而,并非所有药品都有开展PMR的义务,仅有处方药和专利药会受到505(o)条款的约束。而且通常来说,PMR主要针对已经发现风险或有严重风险信号的产品。因而FDA在开展一项PMR之前,必须明确药物相关的不良反应/事件报告,并确认现有药物警戒系统不能达到下述目的:(1)评估上市药品有关的已知严重风险;(2)评估上市药品有关的严重风险信号;(3)识别已知数据所提示的潜在严重风险[10]。基于上述原则,FDAAA授予FDA权力在以下几种情况下,可以向申请人提出PMR:(1)加快程序审批的药品;(2)依据动物药效学研究结果审批的药品;(3)儿童用药的延迟研究;(4)新批准的处方新药及新生物制品的风险评价。
1.3 PMC和PMR的实施情况
一个药品可能拥有多个PMR和PMC,但一般来说,PMR的数量要多于PMC。据FDA官方统计,每年新备案的PMR数量大约是PMC的3~5倍。而根据FDA官方网站最新发布的《药品和生物制品开展上市后要求和承诺情况的报告》统计,截至2015年9月,共有269个申请人提交的856个新药和新生物制品的上市后研究项目(包括PMR和PMC)记录在案,其中包括194个申请人提交的716个新药申请(New drug application,NDA)、74个申请人提交的140个生物制剂许可申请(Biologic license application,BLA)项目。
如上文所述,美国FDA对PMR和PMC的约束力有所不同,在一定程度上导致两者在开展过程中的依从情况存在一定差异。总体来看,虽然FDA记录的PMR项目数量要远高于PMC,监管难度也较PMC更大,但PMR整体的依从水平要高于PMC,主要表现在研究按预定进度开展的比例相对较高、延期比例较低,详见表1(数据截至2015年9月)。

表1 PMR和PMC的开展情况(%%)Tab1 The progress of PMR and PMC(%%)
2 美国药品上市后研究的监管
如上文所述,美国药品上市后研究可分为PMR和PMC两种。但总体来看,FDA对PMR和PMC的监管方式基本一致,所涉及的监管要素、流程和职责分配存在较多重合和相似之处。鉴于此,下文分析美国药品上市后研究的监管方式时,将不针对PMR和PMC作分别阐述,仅在两者存在差异时予以区分。
2.1 监管过程中的关键要素
2.1.1 监管主体在美国上市销售的药品,其上市后研究的监管主要由药品评价和研究中心(Center for Drug Evaluation and Research,CDER)下属的新药办公室(Office of new drug,OND)负责,监测则由流行病学办公室(Office of Surveillance and Epidemiology,OSE)辅助完成[11]。具体来说,一般由来自OND新药评审团队的专业评审员和管理人员完成具体监督和项目评价工作,而这些人员依照其职责设置,可以分为开展具体工作的PMR/PMC的制订协调员、评审员、项目管理员、跟踪协调员、质量管理员等,以及负责整体协调和管理的项目管理经理、评审管理员[12]。
2.1.2 关键文件在药品上市后研究方案制订和开展的过程中,FDA和申请人之间所涉及的关键文件按照其作用可分为两类:一类是帮助FDA和申请人达成研究协议的文件,包括FDA完成审评后起草的“PMR/PMC草案”和最终发送给申请人的“PMR/PMC通知件”;另一类是用于对已确定的研究进行过程跟踪和监督的文件,一般由申请人按规定时间向FDA提交,包括研究的“年度状态报告”“最终报告”和“完善PMR/PMC的补充申请文件”等。
2.1.3 重要时间节点为了跟踪某药品上市后研究的完成进度,并监督申请人在规定时间内完成相关研究工作,FDA通常会要求申请人在研究进度计划和研究申请信中明确提交相关材料的特定节点日期,并将其作为PMR/PMC协议内容的一部分。这些时间节点可以为某一个具体时间,如最终研究协议提交日期、最终协议提交日期或最终研究报告提交日期;也可以为“距前一个节点的时段”,如“最终协议提交日期后的第n个月完成研究”“研究实际完成后的第n个月内提交最终研究报告”等。
2.2 监管流程
从美国药品上市后研究的定义和基本要求可知,FDA的相关监管工作主要可分为两个部分:(1)药品上市前,与申请者协定研究内容,制订研究草案;(2)药品上市后,监督申请者按计划提交研究报告,审核其是否达到要求。
2.2.1 制订研究草案PMR或PMC研究草案的制订工作一般在NDA审评基本完成后开始,至FDA向申请者发送审批件(Approval letter)时正式结束。这一阶段相关监管人员的主要职责为:①明确药品是否有必要开展PMR或PMC;②制订PMR或PMC研究草案,通过沟通交流与申请人就研究内容和进度安排达成一致;③将最终协定的研究方案告知申请人。监管中的方案制订流程见图1。
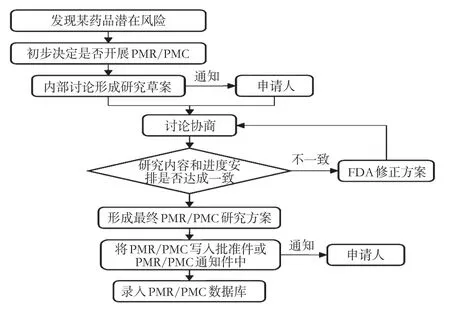
图1 FDA对药品上市后研究监管中的方案制订流程Fig1 Protocol development flow of FDA’s supervision on drug post-marketing research
2.2.2 审核研究报告申请人按协议研究方案开展药品上市后研究后,需要按研究进度计划定期向FDA提交研究报告。在这个过程中,FDA相关监管人员的主要职责在于:①跟踪研究过程,督促申请人按期提交研究报告;②审核研究报告的真实性和科学性,及时发现问题并要求申请人补充材料;③确认已有研究是否实现了证实药品安全性和有效性的目的。监管中的报告审核流程见图2。

图2 FDA对药品上市后研究监管中的报告审核流程Fig2 Report review flow of FDA’s supervision on drug post-marketing research
2.3 配套监管系统
如上文所述,FDA与申请者达成一致的药品上市后研究协议后,PMR/PMC跟踪协调员会将药品的基本信息和最终确定的研究内容登记到PMC/PMR数据库中,并在未来的跟踪和监督过程中,以数据库记录的研究报告提交情况为依据,确定研究的进展状况。
PMR/PMC数据库是一个对外公开的数据库,公众和企业可以通过检索此数据库,了解FDA批准的每个药品的PMR、PMC的基本信息和实施情况,包括申请者信息、产品信息、PMR和PMC内容描述、年度研究报告提交情况以及“当前状态”等。其中,“当前状态”一栏是FDA判定某药品上市后研究实施情况的重要依据,只有达到“已完成”或“已免除”状态,才表明该药品的上市后研究工作全部完成。此外,FDA对PMR/PMC数据库实施动态管理,数据库内公布的数据会在每年的1月、4月和10月定期更新。PMR/PMC数据库中的7种“当前状态”见表2[13]。

表2 PMR/PMC数据库中的7种“当前状态”Tab2 7 kinds of“current condition”in PMR/PMC database
2.4 强制措施
对于PMR,由于其本身具有FDAAA法案赋予的义务性,因而申请者未按要求完成相关研究将被视为违法行为,并会因此受到法律处罚。FDAAA 505(p)条款授权FDA,除非申请人有能够说服FDA监管人员的充分理由,否则可以依照法律对其采取以下几种处罚措施[14]:(1)限制相关药品的州际间贸易;(2)指控相关药品的标签错误;(3)民事罚款。
而对于PMC,虽然此类研究不属于法定义务,未在预定时限内按方案完成研究的申请者不会依法受到行政处罚或民事罚款。但对于此类情形,《美国联邦行政法典》CFR21(Code of Federal Regulations)第9章第356b(e)条提出,FDA局长可以要求申请者通知执业医师,该药厂生产、销售的药品未能完成上市后研究,以至于可能无法解释该药品的某些临床获益情况和安全性问题。FDA将在官方网站上发布声明,告知公众某药品的申请者未按要求完成上市后研究,并说明未达到协议要求的、未完成研究的原因。
3 美国上市后研究制度对我国的启示和建议
通过研究发现,FDA依靠完备、明确的法律条款,充分发挥了自身对药品上市申请者的指导作用和监管职能,实现了对药品上市后研究的科学、规范监管。在此基础上,还通过专有数据库实现了对药品上市后研究状态的实时追踪,督促企业按照研究计划完成研究[15]。这对于完善我国药品Ⅳ期临床制度,充分发挥Ⅳ期临床试验在提高药品安全性、有效性方面的作用,具有一定启示和借鉴意义。
3.1 转换监管思路,充分发挥政府的引导和监督作用
在药品监管方面,我国国家食品药品监督管理总局(CFDA)与FDA的监管理念和监管方式存在较大差异,药品上市后的安全监管也不例外。从美国药品上市后研究的监管流程可以看出,FDA无论在上市后研究协议的制订还是执行过程中,都会邀请申请人参与其中,并注重与申请人的沟通交流,从而保证上市后研究设计良好、执行严格。
对此,笔者认为,监管者应当意识到企业是产品的第一责任人,政府在药品监管过程中所扮演的应是引导者和监督者的角色,而非包揽所有责任。因而,在药品Ⅳ期临床监管制度的制度设计中,应充分考虑监管部门和申请者之间的责任分配。具体来说,即建议药品监管部门允许申请者参与试验内容和试验进度的制订,同时,对申请者提出的科学、合理的修正意见予以考虑和采纳。而当药品上市后,CFDA负有告知申请者的主动报告义务,并以此为纽带保持审评团队与申请人之间的联系,持续指导和监控Ⅳ期临床研究的开展。
3.2 加强药品上市前后监管的衔接,制订特色化Ⅳ期临床方案
无论是上市后的PMC还是PMR,FDA都是基于NDA审评情况,对每个药品制订特色化上市后研究方案,并允许申请人根据自身情况与FDA共同修正。这种做法相比于我国依照药品注册分类,以“一刀切”的方式管理药品Ⅳ期临床试验更加科学、有效。我国应借鉴美国对PMR和PMC的分类管理,充分考虑药品本身的风险,以及申请者提交的NDA材料中关于药品安全性和有效性的证据,以此为依据,有针对性地要求或建议企业开展Ⅳ期临床试验,并制订内容明确、进度安排合理的研究方案。
3.3 建立Ⅳ期临床试验数据管理系统,加强全过程监管
数据管理系统是现代化管理的重要工具,高效率、高质量的管理离不开该工具。Ⅳ期临床试验周期较长、过程复杂,构建相应的数据库及数据管理系统对实施高效跟踪和管理非常必要。借鉴美国PMR/PMC数据库的管理模式,我国CFDA可建立类似平台,对企业每年递交的临床Ⅳ期临床试验报告进行存储,并跟踪发布各品种药品临床Ⅳ期进展及结果的相关信息,以实时关注药品临床Ⅳ期的实施情况。基于该数据库的跟踪监测情况,CFDA可每年汇总并撰写概况报告,总结年度Ⅳ期临床试验的安全情况;同时,数据库中记录的时间节点能够帮助监管部门及时发现、纠正未按规定开展Ⅳ期临床试验的情况,提高药品上市后安全监管的效果。
4 讨论
药品上市所依据的药学、非临床和临床研究往往存在一定固有缺陷。因此对于大多数药品,尤其是首次上市的新药来说,上市后继续在更广泛的人群中开展药品安全性、有效性研究往往是必要的。我国长期以来“重审批、轻监管”,因而对药品上市后研究一直缺乏关注和管理。然而随着药品监管部门逐渐重视“过程监管”,以及相关机构改革和制度改革的同步开展,我国应借鉴美国对药品上市后研究的这种“事前制订计划,事中动态追踪,事后依法处理”的监管方式,以及将药品监管贯穿药品上市后整个周期的思路,为我国完善Ⅳ期临床试验制度和加强药品上市后的风险管理提供重要参考。
[1] 赵建中,谢松梅,杨进波,等.不同国家药品上市后研究管理现状比较[J].中国新药杂志,2014,23(22):2589-2592.
[2] 向秋静,叶桦.关于国外开展药品上市后再评价相关制度的分析[J].中国药事,2016,30(4):406-410.
[3] 刘璐,温宝书,黄清竹,等.Ⅳ期临床试验管理体制的研究探讨[J].中国新药杂志,2010,19(17):1503-1507.
[4] 董铎,孙利华,王丹.美国FDA关于企业开展药品上市后研究和临床试验指南[J].中国新药杂志,2011,20(9):960-962.
[5] FDA.Food and drug administration amendment act[EB/OL].[2016-11-15].https://www.fda.gov/regulatoryinfor-mation/lawsenforcedbyfda/significantam end ments to the fdcact/food and drugadministratio nam end mentsact of2007/default.htm.
[6] 刘平羽.国外药品上市后再评价制度简介[J].上海医药,2004,25(5):208-210.
[7] 孙新欣.美国新药上市后定期汇总报告的研究[J].上海医药,2013,34(21):44-47.
[8] 任经天,吴晔,程鲁榕.美国药品上市后研究承诺简介[J].药物流行病学杂志,2005,14(2):97-100.
[9] 奚晓云,李国芬.美国、欧盟和日本药物警戒法规体系简介[J].药物流行病学杂志,2010,19(10):587-591.
[10] 郭晓昕,杜晓曦.药品风险信号的发现与上市后研究[J].中国临床药理学杂志,2011,27(8):634-641.
[11] 董铎,刘翠丽.美国药品生产企业上市后监测制度研究及启示[J].中国药物警戒,2013,10(8):456-459、463.
[12] FDA.Procedures and responsibilities for developing post market in grequirement sandcom mitments[EB/OL].[2016-11-10].http://101.96.10.43/www.fda.gov/downloads/abooutfda/centers offices/office of medical product sandtobacco/cder/manual of policies procedures/ucm120877.
[13] FDA.Postmarketing requirements and commitments:frequently asked questions(FAQ)[EB/OL].[2016-11-08].http://www.fda.gov/drugs/guidance compliancere gulatory in for mation/post-market in gpha seivco mmitments/ucm-070766.htm.
[14] FDA.Notice to industry:post marketing requirements post marketing studies and clinical trials[EB/OL].[2016-11-08].http://www.fda.gov/drugs/guidance compliancere gulatory in for mation/ucm292758.htm.
[15] 单爱莲,蒋玉凤.新药Ⅳ期临床试验与药品上市后再评价的异同点以及存在的问题[J].中国临床药理学杂志,2014,30(5):387-390、396.
Supervision System on Drug Post-marketing Research in America and Its Enlightenments to China
LUO Xueyan,LAI Han,CHEN Shaocheng,LI Jun(Dept.of Biology and Chemical Engineering,Chongqing University of Education,Chongqing 400067,China)
OBJECTIVE:To put forward suggestions for improving the supervision of phaseⅣclinical trials in China.METHODS:According to summarizing the post-marketing research in America,FDA’s supervision(including key elements,supervision flow,auxiliary supervision system and enforcement measures)for drug post-marketing research in America was analyzed,and suggestions for the supervision of phaseⅣclinical trials in China was put forward.RESULTS&CONCLUSIONS:The drug post-marketing research in America included post-marketing commitment research(PMR)and post-marketing requirement research(PMC).The key elements included supervision subjects(dealt by Office of New Drugs affiliated to Drug Evaluation and Research Center),key document(including the documents helping FDA and applicants reached a research agreement,and documents for process tracking and supervision in identified studies)and important time node.The supervision flow included developing drafts and reviewing reports.FDA had established PMC/PMR database,which was used as auxiliary supervision system,and relevant enforcement measures were respectively developed for PMC and PMR.Relevant supervision departments in China should converse the supervision ideas,give full play to the government’s guidance and supervision,enhance the connection of supervision between pre-and post-marketing,specially develop phaseⅣclinical program,establish system for phaseⅣclinical trial data,enhance whole process supervision,draw lessons from“pre-process plan,dynamic tracking in the process,and post-process decision according to law”of FDA to improve the supervision of phaseⅣclinical trials in China.
Post-marketing commitment research;Post-marketing requirement research;Supervision;PhaseⅣclinical trials;America
R951
A
1001-0408(2017)31-4330-05
DOI 10.6039/j.issn.1001-0408.2017.31.03
重庆市教委2015年度科学技术研究项目:基于“三医联动”模式下的药品采购制度研究——以重庆市为例(No.KJ1501417);重庆第二师范学院2015年度校级科研项目:基本医疗保险药品价格谈判机制研究——以重庆市为例(No.KY201511B)
*讲师。研究方向:药品政策法规、医疗保障政策。电话:023-86388609。E-mail:375597241@qq.com
#通信作者:讲师。研究方向:药品流通领域政策法规。电话:023-86388609。E-mail:2474912362@qq.com
2017-01-20
2017-08-14)
(编辑:刘明伟)
