反式-1-氯-3,3,3-三氟丙烯的制备研究进展
2017-11-09曾纪珺韩升唐晓博赵波郝志军张伟吕剑
曾纪珺,韩升,唐晓博,赵波,郝志军,张伟,吕剑
(1氟氮化工资源高效开发与利用国家重点实验室,陕西 西安 710065;2西安近代化学研究所,陕西 西安 710065)
反式-1-氯-3,3,3-三氟丙烯的制备研究进展
曾纪珺1,2,韩升1,2,唐晓博1,2,赵波1,2,郝志军1,2,张伟1,2,吕剑1,2
(1氟氮化工资源高效开发与利用国家重点实验室,陕西 西安 710065;2西安近代化学研究所,陕西 西安 710065)
反式-1-氯-3,3,3-三氟丙烯[HCFO-1233zd(E)]的臭氧消耗潜值(ODP)为0.00024,温室效应潜值(GWP)为7.0,环保性能优良,已被国际社会认定为第四代氟代烃发泡剂,应用前景广阔。本文对现有HCFO-1233zd(E)制备方法进行综述,通过比较各合成路线优缺点,提出以HCC-240fa及其衍生物为原料的合成路线最具工业化前景。同时,对制备工艺中涉及的HCFO-1233zd(E)与HF和副产物共沸分离问题进行了分析总结。最后指出高效的氟化催化剂和绿色的分离工艺是未来HCFO-1233zd(E)工业化开发的研究重点。
反式-1-氯-3,3,3-三氟丙烯;发泡剂;合成;分离
硬质聚氨酯泡沫塑料(简称硬泡)具有热导率低、耐水性好、比强度高等优点,广泛用作冰箱、冷柜及冷藏集装箱的保温层、建筑保温板材及各类管道设备保温材料。发泡剂是制造硬泡的关键助剂,其种类和用量直接影响着硬泡泡沫的物理力学性能、绝热保温效果和生产成本等[1]。氟代烃作为一种物理发泡剂,具有沸点低、发泡效率高、发泡稳定、与多元醇及聚氨酯基体不发生化学变化、能够良好互溶等特点,特有的隔热性能使其成为保温发泡材料的首选[2-3]。
受环境问题的驱动,氟代烃发泡剂已经发展了三代[4-5]。第一代发泡剂三氯氟甲烷(CFC-11)会破坏大气臭氧层,国际社会已淘汰。第二代发泡剂1,1-二氯-1-氟乙烷(HCFC-141b)为过渡性消耗臭氧层物质(ODS)替代品,属于《蒙特利尔议定书》规定淘汰的物质,发达国家在2004年完成淘汰,我国已于2013年开始逐步削减HCFC-141b的产能,预计在2030年完全停止生产和使用。第三代发泡剂1,1,1,3,3-五氟丙烷(HFC-245fa)的大气臭氧消耗潜值(ODP)为0,不破坏大气臭氧层,是目前国际上主要使用的物理发泡剂,但其温室效应潜值(GWP)为1030,属于《京都议定书》中受关注的温室气体,随着国际社会对温室效应及碳排放日趋严格的控制,HFC-245fa面临受限并淘汰的现实[6]。
近年来,在第四代氟代烃发泡剂的研发过程中,反式-1-氯-3,3,3-三氟丙烯[HCFO-1233zd(E)]以其优异的性能得到一致的认可[7],2016年8月底我国环境保护对外合作中心(FECO)发布的《氢氯氟烃(HCFCs)重点替代品推荐目录(第一批)》)中,E-HCFO-1233zd作为HCFCs发泡剂(HCFC-141b)的推荐替代品位列名单中。HCFO-1233zd(E)的ODP为0.00024,GWP为7.0,环保性能优良,毒性低,常态下不燃,使用安全,采用HCFO-1233zd(E)发泡体系合成的硬泡综合性能良好,绝热性能优异,能够满足绝热保温行业的需求。
依据起始原料的不同,文献报道HCFO-1233zd(E)的合成路线大致有6条,本文对现有制备方法进行评述,对比各类合成路线的优缺点,并对不同路线分离工艺中的共性问题进行分析,以期为工业化路线的选择和新路线的开发提供有益的思路。
1 HCFO-1233zd(E)的制备方法
1.1 以HCC-240fa及其衍生物为原料
以1,1,1,3,3-五氯丙烷(HCC-240fa)及其衍生物,包括1,1,3,3-四氯-2-丙烯(HCC-1230za)、HFC-245fa、1,3,3,3-四氟丙烯(HFO-1234ze)、1,1,1-三氟-3,3-二氯丙烷(HCFC-243fa)和3-氯-1,1,1,3-四氟丙烷(HCFC-244fa)等为起始原料,通过气相(或液相)氟化、加成、氯化氢化、脱卤化氢等反应,一步或多步合成HCFO-1233zd,如图1。

图1 以HCC-240fa及其衍生物合成HCFO-1233zd(E)路线示意图

图2 HCC-240fa氟化合成HCFO-1233zd
1.1.1 以HCC-240fa为原料
气相或液相氟化HCC-240fa可以一步得到HCFO-1233zd(图2),该路线简洁,具备较高的工业价值,研究的关注点是氟化催化剂。
常规的气相氟化催化剂,如Cr2O3[8]、CrCl3/Al2O3[9]、氟化氧化铝[10]、Cr-Ni/AlF3[11]等均可催化气相氟化HCC-240fa,获得100%的转化率,关键是HCFO-1233zd的选择性和催化剂寿命。兰兹等[11]以Cr-Ni/AlF3为催化剂,在管式反应器中气相氟化HCC-240fa合成HCFO-1233zd,反应温度250℃,接触时间2s,HF/HCC-240fa摩尔比为14∶1,HCC-240fa转化率为100%,HCFO-1233zd的选择性为82%(E/Z=7.3),主要副产物是过度氟化物,包括8.2%的HFO-1234ze和8.5%的HFC-245fa。适当降低反应温度可以提高HCFO-1233zd的选择性,避免过度氟化产物的生成,将反应温度降至150℃,接触时间5.8s,HCFO-1233zd的选择性提高至91.4%,副产物HFO-1234ze和HFC-245fa的选择性分别降至0.8%和3.9%。为了延长氧化铬基催化剂的寿命,可以在反应过程中连续送入少量的氧气或氯气。默克尔等[12]通过在进料中引入空气(O2/HCC-240fa摩尔比0.032∶1),在反应过程中同时完成催化剂的再生活化,带走反应热,将氧化铬基催化剂寿命延长到最少两倍。
使用常规的氧化铬基氟化催化剂气相氟化HCC-240fa存在HCFO-1233zd选择性不高、催化剂寿命短等问题,因此有必要开发出低温高活性催化剂,避免高温造成的催化剂表面快速结炭以及深度氟化副产物的生成。低温氟化催化剂通常为“高价金属”卤化物负载型催化剂[13],所谓“高价金属”是指氧化价态至少大于4的金属元素,如Sb、Ta、Mo、V、Ti、Sn和Nb等。吕剑等[14]报道了将锑、钽、铌、钛、锆、钼、钒或锡等高价金属化合物负载在氧化铝、三氧化二铬、氟化铝或氟化镁等载体,制得低温型氟化催化剂,可将气相氟化温度降至150~180℃,远低于氧化铬催化剂所需的200~350℃。优选使用5%的Sn4+/AlF3催化剂,反应温度150℃,接触时间2s,HF/HCC-240fa摩尔比为8∶1,HCC-240fa转化率为100%,HCFO-1233zd的选择性为98.2%。QUAN等[15]报道了SbCl5/PAF(大孔氟化铝)和SbCl5/PCrF(大孔氟化铬)负载型催化剂,研究结果表明,HCFO-1233zd的选择性随着SbCl5负载量的增加而增加,优选使用50%的SbCl5/PCrF催化剂,HCFO-1233zd的选择性为98.8%(E/Z=9.98)。
工业上HCC-240fa一般由四氯化碳与氯乙烯调聚反应制得,反应过程中使用铁配合物为催化剂,并加入N,N-二甲基乙酰胺、乙腈、2-氨基乙腈、N,N-二甲基甲酰胺或六甲基磷酰三胺等有机溶剂。日比野泰雄等[16]发现合成HCC-240fa时残余的微量铁配合物和有机溶剂会降低HCC-240fa的氟化转化率和气相氟化催化剂寿命,并造成装置结垢、腐蚀,需通过水洗、吸附等手段将残余的铁络合物和有机溶剂降至100μL/L以下,以满足气相氟化反应的原料要求。
波克罗夫斯基等[17]报道了不使用催化剂,在釜式反应器中液相氟化HCC-240fa合成HCFO-1233zd,110℃、1.72~2.42MPa(G)下反应9.5h,HCC-240fa转化率为96.8%,HCFO-1233zd(E)的收率为36.2%,HCFO-1233zd(Z)的收率为1.48%,主要副产物为初级氟化产物CFCl2CH2CHCl2(HCFC-241fa,收率为38.7%)。为了提高HCFO-1233zd(E)的收率,波克罗夫斯基提出了串联反应釜工艺,采用3个带有附属精馏塔的连续搅拌式反应器,向第一反应器中连续进料,HF/HCC-240fa摩尔比为15∶1,第一、二和三反应器的操作温度分别为140℃、135℃和130℃,操作压力分别为2.76MPa、2.58MPa和2.41MPa,持续将反应釜底部中间氟化产物送至下一个反应器,采用此工艺可在第三反应器实现100%的HCC-240fa转化率和95%的HCFO-1233zd(E)收率。
液相氟化可以加入路易斯酸催化剂,比如TiCl4、SnCl4、SbCl5或TaCl5等,提高HF体系的酸性,促进反应。汪海有等[18]以TiCl4为催化剂,85℃、0.83MPa(G)下液相氟化HCC-240fa,HCFO-1233zd(E)的收率为86.4%,HCFO-1233zd(Z)的收率为1.5%,主要副产物是过度氟化物HCFC-244fa(收率5.5%)和HFO-1234ze(收率4.2%)。若使用酸性更强的SbCl5或TaCl5[16]为催化剂,则只能得到少量的HCFO-1233zd,主要产物是深度氟化物HFC-245fa和HCFC-244fa,以及低聚物。
HCC-240fa与HF不互溶,可以加入适量的相转移催化剂,提高液相氟化反应速率。汪海有等[19]报道了以I–、Br–、Cl–、F–和(HF)nF–(n=1.0~4.0)为阴离子的一类离子液体为相转移催化剂催化液相氟化HCC-240fa合成HCFO-1233zd,在相同的反应温度下,加入质量分数2.4%的EMImCl(EMIm表示1-乙基-3-甲基咪唑鎓)后,反应时间由不加前的10h缩短至8h,HCFO-1233zd(E)收率由35.5%提高至38.8%。童雪松等[20]以季鏻盐或季铵盐为相转移催化剂,在相同的反应温度下,加入质量分数0.8%的甲基三辛基氯化铵后,反应时间由不加前的9.5h缩短至5h,HCFO-1233zd(E)收率由35.5%提高至38.1%。上述报道表明加入相转移催化剂可以缩短反应时间,但是对HCFO-1233zd(E)收率提高有限。
1.1.2 以HCC-1230za为原料
HCC-1230za可以由HCC-240fa液相催化或高温气相脱氯化氢合成而得,收率一般为90%~95%[21]。气相或液相氟化HCC-1230za一步得到HCFO-1233zd(E)(图3),相比HCC-240fa,HCC-1230za具有更高的氟化反应活性。
埃尔斯海夫[22]报道了在氧化铬催化剂存在下,HCC-1230za气相氟化合成HCFO-1233zd,HCFO-1233zd的选择性与反应温度有直接关系,当反应温度为150℃、接触时间8s、HCFO-1233zd的选择性为93.9%、反应温度提高至200℃时,接触时间7s,HCFO-1233zd的选择性降至53.1%,而过度氟化产物HFC-245fa的选择性由1.1%增加到42.5%。

图3 HCC-1230za氟化合成HCFO-1233zd
使用常规的路易斯酸(如TiCl4或SbCl5)催化液相氟化HCC-1230za主要得到低聚产物。一种解决思路是不使用催化剂,MAHER等[23]报道了在无催化剂存在下,高HF/HCC-1230za的摩尔比(166∶1)、70℃、1.17MPa下反应3h,HCC-1230za的转化率100%,HCFO-1233zd选择性为97%,反应产物中检测不到低聚物。为了验证无催化液相氟化HCC-1230za的工艺可行性,PIGAMO等[24]在1L的不锈钢高压釜中开展了连续氟化HCC-1230za的研究,预加入300g HF,随后将反应器温度升至91~92℃,连续通入40g/h的HCC-1230za和44g/h的HF 200h,HCC-1230za的转化率100%,HCFO-1233zd(E)的收率为91%,HCFO-1233zd(E)的时空收率为0.68mol/h·L,低聚物的生成率为1.8%,研究结果表明无催化液相氟化HCC-1230za路线具有较好的工业应用价值。另外一种解决思路是使用更弱酸性的催化剂,陈冰等[25]报道了以三氟甲磺酸或三氟乙酸为催化剂,50℃、2.76MPa下反应1h,HF与HCC-1230za摩尔比为160∶1,HCC-1230za的转化率100%,HCFO-1233zd的收率为87%,低聚产物的生成率为1.1%。
1.1.3 以HFC-245fa为原料
浜崎秀男等[26]以氟化氧化铝、Cr/活性炭和Cr/氟化氧化铝等为催化剂,催化HFC-245fa与HCl反应得到HCFO-1233zd和HFO-1234ze的混合产物(图4)。在310℃、N2稀释下,以氟化氧化铝为催化剂,产物比例与HCl/HFC-245fa的摩尔比有关,当HCl/HFC-245fa的摩尔比为5.9时,HCFO-1233zd/HFO-1234ze的选择性比值为13.9,当HCl/HFC-245fa的摩尔比降至2.2时,HCFO-1233zd/HFO-1234ze的选择性比值降为2.0,不过HCFO-1233zd的反式与顺式比无变化,均为8.05。该路线原料HFC-245fa已实现规模化生产,进一步优化催化剂和反应条件可以获得更高的HCFO-1233zd选择性,也可联产HCFO-1233zd和HFO-1234ze,是一条潜在的工业应用路线。

图4 HFC-245fa合成HCFO-1233zd
1.1.4 以HFO-1234ze为原料
MASAMUNE等[27]以AlF3、Zr-Nb/AlF3和CrF3为催化剂,HFO-1234ze与HCl反应合成HCFO-1233zd。以Zr-Nb/AlF3为催化剂,HFO-1234ze(Z)与含2.3% HF的HCl为原料,反应温度310℃,接触时间4.8s,HFO-1234ze(Z)转化率99.5%,HCFO-1233zd(E)选择性84.59%,HCFO-1233zd(Z)选择性10.38%,E/Z比为8.15。受热力学平衡的影响,HFO-1234ze(E)的反应活性低于HFO-1234ze(Z),反应温度为280℃时,HFO-1234ze(E)的转化率只有57.16%,不过将反应温度提高至360℃,HFO-1234ze(E)的转化率可达98.4%。该路线原料HFO-1234ze(E)是新型气体发泡剂和喷雾剂,还未工业化生产,原料价格昂贵导致本路线经济性差,不过将合成HFO-1234ze(E)过程中暂无用途的顺式异构体HFO-1234ze(Z)转化为HCFO-1233zd(E)是一个很好的联产思路。
1.1.5 以HCFC-243fa或HCFC-244fa为原料
以HCFC-243fa或HCFC-244fa为原料,在无机碱或高温气相中脱卤化氢可以一步获得HCFO-1233zd(E)。
HASZELDINE[28]使用固体粉末KOH为碱源,在回流温度下处理HCFC-243fa 12h,HCFO-1233zd的收率仅为16%,继续脱氯化氢产物三氟丙炔的收率为45%。肖恒侨等[29]改进了碱脱HCFC-243fa的方法,使用30%的KOH水溶液为碱源,并加入相转移催化剂四丁基溴化铵,70~80℃、0.2~0.5MPa下反应3.5h,HCFC-243fa的转化率为100%,HCFO-1233zd的收率为96.3%,副产物三氟丙炔只有1.5%。WANG等[30]报道了以HCFC-243fa原料气相脱氯化氢合成HCFO-1233zd的方法,考察了氟化Cr2O3、AlF3、10% FeCl3/活性炭和0.5% Fe/活性炭的催化脱氯化氢活性,其中氟化Cr2O3效果最佳,反应温度250℃,接触时间44.4s,HCFC-243fa转化率为91.0%,HCFO-1233zd(E)选择性为90.6%,HCFO-1233zd(Z)选择性为8.5%,相比液相碱脱法,气相催化法不产生固废(NaCl),更环保。
MERKEL等[31]报道了以金属氯化物负载在活性炭或MgF2为催化剂,HCFC-244fa气相脱氟化氢合成HCFO-1233zd的方法。HCFC-244fa可以同时发生脱氯化氢和脱氟化氢反应,反应的转化率和产物的选择性与金属氯化物(选自KCl、MgCl2、NiCl2、CuCl2、ZnCl2或FeCl3等)和载体(活性炭或MgF2)的酸碱性有关。在350℃、接触时间40.2s的条件下,以10.0% FeCl3/C为催化剂获得了最高的HCFO-1233zd收率(41.0%)。MERKEL等[31]还考察了氟化Cr2O3、AlF3等金属氟化物催化剂的催化效果,在相同的反应条件下,HCFC-244fa转化率为100%,HCFO-1233zd的选择性为74.6%,HFO-1234ze的选择性为20.7%,即使用更强酸性的金属氟化物为催化剂时主要发生脱氟化氢反应。为了提高HCFC-244fa气相脱氟化氢的选择性,HIDEAKI等[32]引入氯化氢作为反应物,HCl/HCFC-244fa摩尔比为9.89,反应温度300℃,接触时间9.98s,HCFC-244fa的转化率为98.85%,HCFO-1233zd的选择性为90.48%(E/Z=8.2),HFO-1234ze的选择性只有0.47%。
原料HCFC-243fa和HCFC-244fa可以由HCC-240fa液相氟化制备,但单一组分选择性不超过70%[33-34],因此本路线应以HCC-240fa为起始原料,两步法联产HCFO-1233zd和HFO-1234ze。
1.2 以HCC-230为原料
ZHAI等[35]以CCl3CHClCHCl2(HCC-230)为起始原料,经氟化、脱氯两步反应合成HCFO-1233zd(图5)。首先以SbCl5为催化剂,液相氟化HCC-230合成CF3CHClCHCl2(HCFC-233da),100℃下反应15h,HCFC-233da收率为38.6%。随后以去离子水为溶剂,HCFC- 233da经锌粉脱氯得到目标产物,HCFO-1233zd(E)的收率为44.4%,HCFO-1233zd(Z)的收率为36.4%。由于原料HCC-230不易得,使用化学计量的锌粉,HCFO-1233zd(E)的总收率只有17.1%,因此该路线工业价值不大。

图5 以HCC-230为原料路线图
1.3 以HCFC-243db为原料
MAHLER等[36]以98%铬/2%钴为催化剂,气相氟化CF3CHClCH2Cl(HCFC-243db)合成HCFO-1233zd,反应温度400℃,HF与HCFC-243db摩尔比为20∶1,HCFC-243db转化率为100%,产物为各种烯烃的混合物,包括2,3,3,3-四氟丙烯(HFO-1234yf)、2-氯-3,3,3-三氟丙烯(HCFO-1233xf)、HFO-1234ze和HCFO-1233zd等,其中HCFO-1233zd选择性只有19.7%。该路线原料HCFC-243db可以由三氟丙烯氯化制备,但是三氟丙烯的高价格以及HCFO-1233zd的低选择性等问题制约了该路线的工业应用。
1.4 以HCFO-1233xf为原料
王博等[39]以HCFO-1233xf为原料,经脱氯化氢、加氯化氢两步反应合成HCFO-1233zd(图6)。首先在500℃下高温裂解HCFO-1233xf合成3,3,3-三氟丙炔,HCFO-1233xf转化率为29.1%,3,3,3-三氟丙炔选择性为88.7%。随后在氟化铬基催化剂存在下3,3,3-三氟丙炔气相加成HCl合成HCFO-1233zd,反应温度320℃,HCl/3,3,3-三氟丙炔摩尔比为4∶1,三氟丙炔转化率为100%,HCFO-1233zd(E)的选择性为69.2%,HCFO-1233zd(Z)的选择性为20.6%。ZHAI等[38]也报道了在氟化Cr2O3存在下3,3,3-三氟丙炔加成HCl主要得到反式HCFO-1233zd,不过当使用CuCl/活性炭为催化剂时,300℃、N2稀释条件下,顺式HCFO-1233zd选择性可达74.1%。

图6 以HCFO-1233xf为原料路线图
王博等[39]还报道了HCFO-1233xf气相异构化一步合成HCFO-1233zd,以氟化铬为催化剂,反应温度350℃,接触时间35s,HCFO-1233xf转化率为50.1%,HCFO-1233zd(E)选择性为64.9%,HCFO-1233zd(Z)选择性为32.3%。
HCFO-1233xf可以由1,1,2,3-四氯丙烯气相氟化制得,主要用于合成第四代制冷剂HFO-1234yf[39],目前HCFO-1233xf还未工业化,而由HCFO-1233xf合成HCFO-1233zd(E)的收率也不高(最高为32.5%),因此该路线商业价值不大。
1.5 以HFC-23为原料
NAIR等[40]设计了数条以CF3H(HFC-23)为起始原料合成HCFO-1233zd的路线,见图7。路线一:以Cu/C为催化剂,300℃下HFC-23与1,2-二氯乙烯发生调聚反应合成HCFO-1233zd,收率在20%~30%;路线二:HFC-23在活性炭催化下气相氯化得到CF3Cl,CF3Cl与氯乙烯调聚合成HCFC-243fa,随后脱氯化氢得到HCFO-1233zd,路线总收率约为55.7%;路线三:HFC-23在活性炭催化下气相氯化得到CF3Cl,CF3Cl与偏氯乙烯调聚合成CF3CH2CCl3(HCFC-233),HCFC-233经脱氯化氢、加氢制得HCFC-243fa,随后HCFC-243fa脱氯化氢制备HCFO-1233zd,路线总收率约为19.9%;路线四:HFC-23在活性炭催化下气相氯化得到CF3Cl,CF3Cl与1,2-二氯乙烯通过Fe/FeCl3/Bu3PO4催化进行1∶1调聚制备HCFC-233da,随后以甲醇为溶剂,50~55℃下HCFC-233da经锌粉(由乙酸/乙酸酐活化)脱氯制得HCFO-1233zd,路线总收率约为36%。
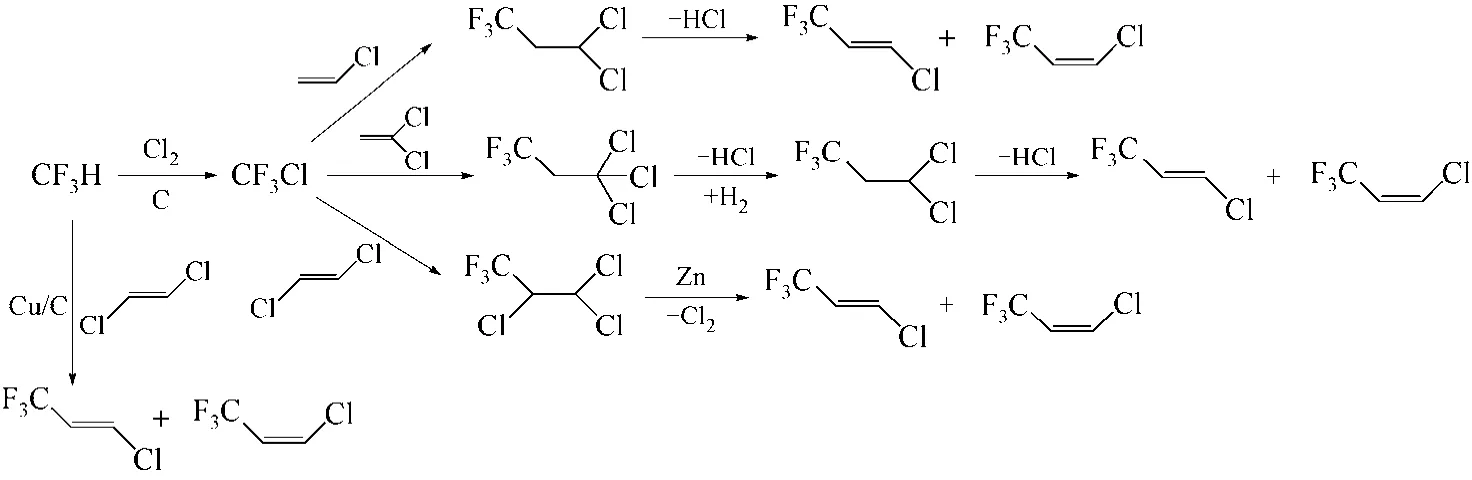
图7 以HFC-23为起始原料的合成路线
HFC-23是合成HCFC-22的副产物,我国每年有大约5400t的量,一般焚烧处理[41],因此该路线为HFC-23的绿色转化提供了一种思路,由于HFC-23和CF3Cl的调聚活性低,路线总收率不高,还需进一步优化。
1.6 以HFC-1243zf为原料
NAIR等[42]设计了3条以三氟丙烯(HFC-1243zf)为起始原料合成HCFO-1233zd的路线(图8)。路线一:HFC-1243zf一步氧氯法。以CuCl2/C为催化剂,450~500℃下HFC-1243zf与氧气和氯化氢以1∶0.5∶2的比例进入固定床反应器,接触时间为5~20s,HCFO-1233zd收率在20%~50%;路线二:氯化碘加成到HFC-1243zf,得到CF3CHICH2Cl,随后气相脱碘化氢得到HCFO-1233zd和HCFO-1233xf的混合物,HCFO-1233zd的总收率约为24%;路线三:在Pd/C催化剂存在下,HFC-1243zf加氢制得三氟丙烷,随后高温气相氯化三氟丙烷得到HCFC-243fa,HCFC-243fa在氟化Cr2O3催化下,400℃、接触时间25~30s,气相脱氯化氢得到HCFO-1233zd,路线总收率约为63%。

图8 以HFC-1243zf为起始原料的合成路线
HFC-1243zf是合成氟硅单体的重要原料之一,已实现工业化生产,但市场规模小,年生产规模仅有数百吨,价格昂贵,限制了该路线的工业化应用。此外,在已报道的3条路线中,HFC-1243zf一步氧氯法是相对绿色的路线,但该路线收率不高,催化剂还需优化。
1.7 HCFO-1233zd(Z)异构化
一般来讲,各路线合成出的HCFO-1233zd普遍为顺反混合物,为了提高反式收率,需将分离出来的顺式HCFO-1233zd异构化为反式HCFO-1233zd。
WANG等[30]考察了氟化Cr2O3、AlF3、10%FeCl3/活性炭和0.5%Fe/活性炭等催化剂的催化异构活性,其中氟化Cr2O3效果最佳,反应温度100℃,接触时间35s,HCFO-1233zd(Z)转化率为68.0%,HCFO-1233zd(E)选择性为99.9%。
冈本觉等[43]发现未干燥处理的氟化氧化铝无催化异构HCFO-1233zd(Z)的活性,将氟化氧化铝在500℃下N2干燥处理3h以上则具有很好的催化活性,反应温度45℃,接触时间192s,HCFO-1233zd(Z)转化率为97%,HCFO-1233zd(E)选择性为99.7%。
1.8 工业路线的选择
从原料来源、技术成熟度、产物收率及产物分离工艺的难易程度等方面分析比较上述合成路线,发现以HCC-240fa及其衍生物为原料合成HCFO-1233zd(E)是最具工业前景的路线。若考虑高选择性地合成HCFO-1233zd(E),那么以HCC-240fa或HCC-1230za原料一步液相或气相氟化是最佳工业路线。另一方面,HFO-1234ze(E)是下一代气体发泡剂和喷雾剂[44],用于替代1,1,1,2-四氟乙烷(R134a)和二氟一氯甲烷(R22)。HFC-245fa是第三代发泡剂,尽管作为发泡剂将被受限和禁用,但目前过渡期仍需要一定的产能,此外还可作为离心机制冷剂替代1,1-二氯-2,2,2三氟乙烷(R123),因此HFO-1234ze(E)和HFC-245fa皆是未来若干年主要的氟代烃产品,若考虑同时联产HFC-245fa或HFO-1234ze(E),那么以HCC-240fa为原料,以HFC-245fa、HCFC-244fa和HCFO-1233zd等为中间产物的多步法是更优选的路线[45-46]。
2 HCFO-1233zd(E)的分离提纯
以HCC-240fa及其衍生物为原料合成HCFO-1233zd(E)工艺一般都使用过量HF作为氟化剂,氟化反应后过量的HF与HCFO-1233zd(E)形成最低共沸物[47],常规精馏难以分离出HCFO-1233zd(E),回收HF。此外,反应产物中一般还包含氟化副产物,比如HFC-245fa、HCFC-244fa和HFO-1234ze(E/Z)等,其中HCFO-1233zd(E)与HFC-245fa形成共沸物,与HFO-1234ze(Z)形成近似共沸物,常规精馏难以获得目标纯度的HCFO-1233zd(E)。为了实现HCFO-1233zd(E)的有效分离,需采用特殊精馏(共沸精馏或萃取精馏)、低温相分、吸附分离和化学吸收等单元操作。
2.1 HF的分离
分离HCFO-1233zd(E)中的HF可以采用浓硫酸吸收、低温相分和共沸精馏耦合法等方法。
浓硫酸吸收法利用HF在浓硫酸中的溶解度远大于HCFO-1233zd(E),低温浓硫酸吸收HF,高温将HF解析出去,浓硫酸循环套用。波克罗夫斯基等[17]采用一套包含填料吸收塔、四氟闪蒸罐和HF回收塔的装置流程实现了HF的回收,以70%的HCFO-1233zd(E)和30%的HF混合物为原料,在填料吸收塔塔顶获得含1.0%HF的HCFO-1233zd(E)产物,在HF回收塔塔顶获得包含少于50μL/L的硫杂质和少于100μL/L水的HF。浓硫酸吸收法中存在含水的氢氟酸和硫酸属于强腐蚀性物质,而回收浓硫酸和HF时必须高温加热,所以该体系腐蚀性大,对设备要求高,泄露风险大。
低温相分与共沸精馏耦合法包含低温相分和共沸精馏两个操作单元[48]。首先低温相分操作,利用低温时HF与HCFO-1233zd(E)不相溶和密度差,形成两相体系。波克罗夫斯基等[17]测定了在-20~0℃范围内HF与HCFO-1233zd(E)的相分情况,温度越低,相分效果越好,以质量比为1∶1的HF与HCFO-1233zd(E)的混合物为对象,–20℃下保持60min,底部为含2.7%HF的有机相,上部为含28.4%HCFO-1233zd(E)的HF相,也即低温相分别获得了上层富HF物流和下层富HCFO-1233zd物流。接着共沸精馏操作利用HF与HCFO-1233zd(E)形成最低共沸物,从而在精馏塔塔顶得到HF与HCFO-1233zd(E)的共沸物,在塔釜得到纯的HF(或HCFO-1233zd(E))。威斯马等[48]采用一套包含相分器、HCFO-1233zd精馏塔和HF回收塔的装置流程实现了HF和HCFO-1233zd的有效分离(图9),以包含64.9%HCFO-1233zd(E)、6.49%HCFO-1233zd(Z)和28.4%HF的共沸混合物为对象,–20℃相分后,上层富HF物流进入HF回收塔,下层富HCFO-1233zd物流进入HCFO-1233zd精馏塔。共沸精馏操作,在HF回收塔的塔釜得到100%的HF,在HCFO-1233zd精馏塔的塔釜得到不含HF的HCFO-1233zd。HF回收塔和HCFO-1233zd精馏塔的塔顶均为含质量分数28.5%HF的共沸物,返回至相分器。低温相分与共沸精馏耦合法物料体系腐蚀性小,使用常规的碳钢或不锈钢设备即可,不过过程涉及低温和气化-液化循环过程,能耗大,因此在流程设计时需要考虑能量耦合,降低能耗。
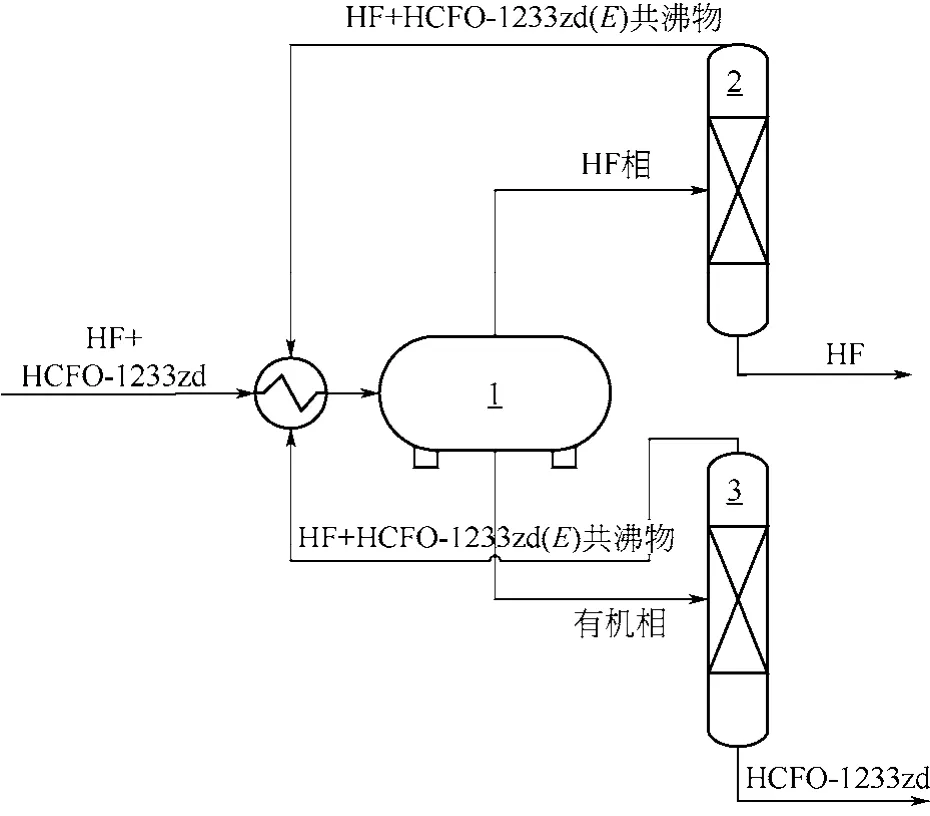
图9 低温相分与共沸精馏耦合法工艺流程
采用浓硫酸吸收、低温相分与共沸精馏耦合法等方法可以有效分离HCFO-1233zd中的HF,但得到的HCFO-1233zd粗品中一般会含有几十到上千μL/L的HF,而HCFO-1233zd产品中酸度要求是小于1μL/L,因此需采用水洗-碱洗工序将HCFO-1233zd粗品洗成中性,这也是绝大部分ODS替代品生产工艺中必要的一个工段。常规碱洗一般使用氢氧化钠水溶液,而HCFO-1233zd粗品中的HCFC-244fa、HCFC-243fa和2-氯-1,1,1,3,3-五氟丙烷(HCFC-235da)等对碱不稳定,会脱氯化氢生成新的烯烃杂质以及三氟丙炔,降低产物收率,而且生成的新烯烃杂质与HCFO-1233zd(E)沸点接近,将带来后续的分离问题。为此,冈本正宗等[49]提出采用pKa为7~11的弱碱,比如碱金属的碳酸盐、碳酸氢盐、磷酸盐、磷酸二氢盐、磷酸氢盐或碳数3~15的叔胺等,代替氢氧化钠水溶液,HF去除率大于99%,同时有效避免了使用强碱分解生成难以分离的烯烃的问题。此外,科卡利等[50]还提出了水洗-浓硫酸洗涤的工艺,避免使用氢氧化钠水溶液除酸生成三氟丙炔的问题,首先水洗将HCFO-1233zd(E)粗品洗涤至近中性,随后浓硫酸洗涤除去残余的氟化氢,并干燥除水,得到不含三氟丙炔的脱酸干燥粗品。
2.2 副产物的分离
除酸后的HCFO-1233zd(E)(沸点18.9℃)粗品一般含有HFO-1234ze(E)(沸点–18.5℃)、HFO-1234ze(Z)(沸点9.2℃)、HFC-245fa(沸点15.3℃)、HCFC-244fa(沸点43℃)和HCFO-1233zd(Z)(沸点39℃)等副产物[51-52],其中HFC-245fa与HCFO-1233zd(E)形成最低共沸物,可以通过共沸精馏以轻组分的形式将其脱除。波克罗夫斯基等[53]使用理论塔板约为30块的精馏塔,在约30psig(1psig=6894.76Pa)的压力下进行间歇精馏操作,得到轻组分馏分和HCFO-1233zd(E)主馏分两个馏分,轻组分馏分色谱分析表明,HFC-245fa与HCFO-1233zd(E)以共沸物的形式从塔顶出去,HCFO-1233zd(E)主馏分的纯度可达99.97%,HFC-245fa含量只有0.0084%。
合成HCFO-1233zd的原料HCC-240fa中一般含有极少量的CCl4,CCl4在氟化过程中会转化为CFCl3(R11)。R11沸点为23.8℃,与HCFO-1233zd(E)沸点接近,形成共沸物,常规精馏难以将两者分开。更为严重的是,R11的ODP为1.0,少量的R11将显著增大HCFO-1233zd(E)产品的ODP,例如,如果HCFO-1233zd(E)被仅仅1000μL/L的R11污染,其ODP将从0.00034增加到0.00134。因此有必要通过特殊手段降低HCFO-1233zd产品中的R11含量。萃取精馏是ODS替代品工艺中分离共沸物的常用手段。萃取精馏通过加入适当选择的萃取剂改变被分离组分间的气液平衡关系,从而使它们变得易于精馏分离,萃取剂的选择是关键。威斯马[54]报道了采用沸点在60~100℃的氯代烃,包括三氯乙烯、四氯化碳、氯仿和甲基氯仿等为萃取剂,选择性的萃取R11,从萃取塔塔顶获得更纯的HCFO-1233zd(E)。使用由两个精馏塔组成的连续萃取精馏操作(图10),采用三氯乙烯为萃取剂,萃取剂与HCFO-1233zd(E)的进料比为1.5,通过萃取操作,萃取塔塔顶HCFO-1233zd(E)产品中R11含量由0.2215%降至0.1131%。
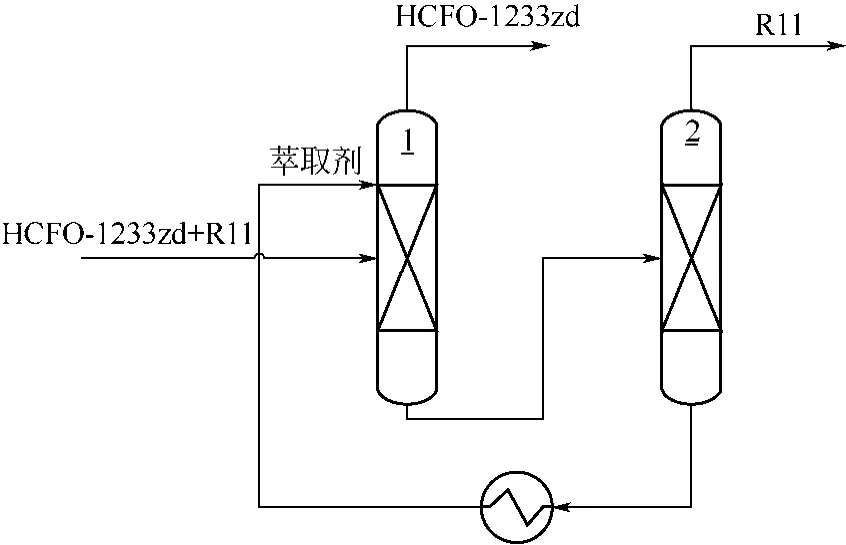
图10 萃取精馏分离HCFO-1233zd(E)和R11工艺流程
在医药、工作流体和热物性测定等领域可能需要超高纯度的HCFO-1233zd(E),常规精馏提纯效率低、能耗大,固体吸附法是一个有效的手段,可以将市售的HCFO-1233zd(E)产品进一步高纯度化。固体吸附法利用多孔性固体吸附剂处理样品,使其中的一种或几种组分,在固体吸附剂表面,在分子引力或化学键力的作用下,被吸附在固体表面,从而达到分离的目的。井村英明等[55]采用X型沸石吸附剂,在0~3℃下带压吸附处理纯度为99.9912%的HCFO-1233zd(E)样品,处理后HCFO-1233zd(E)纯度提高至99.9992%,在精馏中未除净的HFO-1234ze(Z)、HFC-245fa和HFO-1234ze(E)在吸附处理后未检测出,这表明固体吸附法对于HCFO-1233zd(E)的高纯度化是有效的。
3 结语
在已报道制备HCFO-1233zd(E)的众多路线中,具有工业前景的路线是以HCC-240fa及其衍生物为起始原料,适当调整也可以实现同时联产HFC-245fa和HFO-1234ze(E),满足未来对低GWP发泡剂、气雾剂和制冷剂更新换代的需求。高效的氟化催化剂是制备HCFO-1233zd(E)的关键,需加快对气相或液相氟化催化剂的改进性研究:优化低温高活性气相氟化催化剂,提高气相氟化催化剂的寿命,满足工业应用要求;开发酸性可调的液相氟化催化剂,提高液相氟化的转化率和选择性。绿色分离工艺是制备HCFO-1233zd(E)的核心,围绕HCFO-1233zd(E)与HF、近似共沸副产物的难分离问题,需进一步完善分离体系的气液、液液和气固相平衡数据的测定,为分离方法的选择和工业设计提供基础数据。目前,全球只有霍尼韦尔公司、阿科玛公司和中央旭硝子公司有HCFO-1233zd(E)的模试或工业生产装置,国内未见其工业化报道。因此,应加快研究开发力度,尽快形成国产化的HCFO-1233zd(E)工业技术和知识产权保护。
[1] 万元俊,沈晨光,樊陆欢,等. 零ODP聚氨酯泡沫发泡剂替代技术进展[J]. 聚氨酯工业,2017,32(1):1-4.WAN Yuanjun,SHEN Chenguang,FAN Luhuan, et al. Alternative technology progress of polyurethane foam blowing agent with zero-ODP[J]. Polyurethane Industry,2017,32(1):1-4.
[2] 李宇宸,罗振扬. 聚氨酯硬质泡沫用发泡剂的发展现状与HCFC-141b替代面临的挑战[J]. 聚氨酯工业,2014,29(5):1-4.LI Yuchen,LUO Zhenyang. The development of foaming agent for polyurethane rigid foam and the challenge of HCFC-141b substitution[J]. Polyurethane Industry,2014,29(5):1-4.
[3] ENGELS H W,PIRKL H G,ALBERS R,et al. Polyurethanes:versatile materials and sustainable problem solvers for today’s challenges[J]. Angew. Chem. Int. Ed.,2013,52:9422 -9441.
[4] 郑琳飞. 对环境友好的几种聚氨酯泡沫塑料发泡技术介绍[J]. 聚氨酯工业,2016,31(4):44-46.ZHENG Linfei. Some environmental friendly foaming technique of polyurethane foam[J]. Polyurethane Industry,2016,31(4):44-46.
[5] 赵波,陆居有,毛伟,等. 氟代烃类发泡剂的研究进展[J]. 化工进展,2014,33(7):1864-1870.ZHAO Bo,LU Juyou,MAO Wei,et al. Progress in fluorocarbon blowing agent[J]. Chem. Ind. Eng. Prog.,2014,33(7):1864-1870.
[6] 杨春光,杜长明,徐鹤. 基于环境友好型发泡剂的硬质聚氨酯泡沫塑料的性能研究[J]. 中国塑料,2014,28(2):45-50.YANU Chunguang,DU Changming,XU He. Properties of rigid polyurethane foam with environmentally friendly blowing agent[J].China Plastics,2014,28(2):45-50.
[7] 熊丽媛,邢益辉,王金祥. 反式-1-氯-3,3,3-三氟丙烯发泡剂在聚氨酯硬泡中的应用研究[J]. 聚氨酯工业,2016,31(3):26-29.XIONG Liyuan,XING Yihui,WANG Jinxiang. The application of trans-1-chloro-3,3,3-trifluoropropene blowing agent in polyurethane rigid foam[J]. Polyurethane Industry,2016,31(3):26-29.
[8] TONG X S. Vapor phase process for making 1,1,1,3,3-pentafluoropropane and 1-chloro-3,3,3-trifluoropropene:US5710352[P]. 1999-01-27.
[9] YOSHIKAWA S,TAMAI R,SAKYU F,et al. Method for producing1,1,1,3,3-pentafluoropropane:US6198010Bl[P]. 2001-03- 06.
[10] 中田龙夫,青山博一,山本明典. 1,1,1,3,3-五氟丙烷的制备:1206394A[P]. 1999-01-27.TATSUO N,HIROKAZU A,AKINORI Y. Process for producing 1,1,1,3,3-pentafluoropropane:1206394A[P]. 1999-01-27.
[11] 兰兹 A,文德林格 L,勒基米 B. 1-氯-3,3,3-三氟丙烯合成方法及其氟化成1,1,1,3,3-五氟丙烷的方法:1166479A[P].1997-12-03.LANTZ A,WENDLINGER L,REQUIREME B. Synthesis of 1-chloro-3,3,3-trifluoropropene and its fluorination to 1,1,1,3,3-pentafluoropropane:1166479A[P]. 1997-12-03.
[12] 默克尔 D C,波克罗夫斯基 K A,童 X S. 气相生产1-氯-3,3,3-三氟丙烯的催化剂寿命改进:102933532A[P]. 2013-02-13.MERKEL D C,POKROVSKI K A,TONG X S. Catalyst life improvement for vapor phase manufacture of 1-chloro-3,3,3-trifluoropropene:102933532A[P]. 2013-02-13.
[13] YASUO H,PYOUICHI T,SHOUZOU K. Method for producing fluorinated propane:EP19990103578[P]. 1999-09-01.
[14] 吕剑,张伟,王博,等. 1-氯-3,3,3-三氟丙烯的制备方法:101028994B[P]. 2011-05-25.LV J,ZHANG W,WANG B,et al. Method for producing 1-chlorine-3,3,3-triflupropylene:101028994B[P]. 2011-05-25.
[15] QUAN H D,YANG H E,MASANORI T,et al. Preparation of 1,1,1,3,3-pentafluoropropane (HFC-245fa) by using a SbF5-attached catalyst[J]. Journal of Fluorine Chemistry,2007,128:190-195.
[16] 日比野泰雄,吉川悟,佐久冬彦. 1-氯-3,3,3-三氟丙烯的制造方法:104039745A[P]. 2014-09-10.HIBINO Y,YOSHIKAW S,SAKYU F. Production method for 1-chloro-3,3,3-trifluoropropene:104039745A[P]. 2014-09-10.
[17] 波克罗夫斯基 K A,默克尔 D C,童雪松. 生产反式-1-氯-3,3,3-三氟丙烯的连续低温方法:103189339A[P]. 2013-07-03.POKROVSKI K A,MERKEL D C,TONG X S. Continuous low-temperature process to produce trans-1-chloro-3,3,3-trifluoropropene:103189339A[P]. 2013-07-03.
[18] 汪海有,默克尔 D C. 联合生产1,1,1,3,3-五氟丙烷、反式-1-氯-3,3,3-三氟丙烯和反式-1,3,3,3-四氟丙烯的集成方法:103476736A[P]. 2013-12-25.WANG H Y,MERKEL D C. Integrated process toco-produce 1,1,1,3,3-pentafluoropropane,trans-1-chloro-3,3,3-trifluoropropene and trans-1,3,3,3-tetrafluoropropene:103476736A[P]. 2013-12-25.
[19] 汪海有,童雪松. 用于在离子液体中制备1-氯-3,3,3-三氟丙烯的方法:105849072A[P]. 2016-08-10.WANG H Y,TONG X S. Process for producing 1-chloro-3,3,3-trifluoropropene in an ionic liquid:105849072A[P].2016-08-10.
[20] 童雪松,汪海有,默克尔 D C,等. 使用相转移催化剂制备1-氯-3,3,3-三氟丙烯的方法:105026347A[P]. 2015-11-04.TONG X S,WANG H Y,MERKEL D C,et al. Process for the preparation of 1-chloro-3,3,3-trifluoropropene using a phase transfer catalyst:105026347A[P]. 2015-11-04.
[21] OWENS S,JACKSON A,SHARMA V,et al. Methods and materials for the preparation and purification of halogenated hydrocarbons:US20030028057Al[P]. 2003-02-06.
[22] 埃尔斯海夫 M Y. 1,1,3,3-四氯-2-丙烯的气相氟化:98109486.4[P].2004-01-28.ELSHEIKH M Y. Gas phase fluorination of 1,1,3,3-tetrachloro-2-propene:98109486.4[P]. 2004-01-28.
[23] MAHER Y E,ANNE P,SRI R S. Process for the manufacture of hydrochlorofluoroolefins:US8987535B2[P]. 2015-03-24.
[24] PIGAMO A,WISMER J,COLLIER B,et al. E-1-chloro-3,3,3-trifluoropropene production process from 1,1,3,3- tetrachloropropene:US9255045B2[P]. 2016-02-09.
[25] 陈冰,博默 M S,埃尔斯海夫 M Y. 1230za的液相催化氟化:00133009.8[P]. 2001-07-05.CHEN B,BOMER M S,AIRSHAIKAN M Y. Liquid phase catalytic fluorination of 1230za:00133009.8[P]. 2001-07-05.
[26] 浜崎秀男,日比野泰雄. 氟化丙烯的制造方法:102149659B[P].2013-07-31.HIDEO H,YASUO H. Process for producing fluorinated propene:102149659B[P]. 2013-07-31.
[27] MASAMUNE O,HIDEAKI I,NAOTO T. Method for production of 1-chloro-3,3,3-trifluoropropene:US20140005447Al[P]. 2014-01-02.
[28] HASZELDINE R N. Reactions of fluorocarbon radicals. part v.alternative syntheses for trifluoromethylacetylene (3,3,3-trifluoropropyne),and the influence of polyfluoro-groups on adjacent hydrogen and halogen atoms[J]. J. Chem. Soc.,1951:2495-2504.
[29] 肖恒侨,韩国庆,徐卫国,等. 1-氯-3,3,3-三氟丙烯的制备方法:103864570A[P]. 2014-06-18.XIAO H Q,HAN G Q,XU W G,et al. Preparation method of 1-chloro-3,3,3-trifluoropropene:103864570A[P]. 2014-06-18.
[30] WANG H Y,TONG X S. Process for producing trans-1233zd:US8653309B2[P]. 2014-02-18.
[31] MERKEL D C,WANG H Y,POKROVSKI K A,et al. Integrated process toco-produce trans-1-chloro-3,3,3-trifluoropropene,trans-1,3,3,3-tetrafluoropropene,and 1,1,1,3,3-pentafluoropropane:US8648221B2[P]. 2014-02-11.
[32] HIDEAKI I,NAOTO T. Method for production of 1-chloro-3,3,3-trifluoropropene:US20140005446Al[P]. 2014-01-02.
[33] MOSCOE J J. Process for the preparation of 1,1-dichloro-3,3,3-trifluoropropane:US7067705B2[P]. 2006-06-27.
[34] WILMET V,JANSSENS F. Hydro-fluorination of chlorinated hydrocarbons:US6362383B1. 2002-03-26.
[35] ZHAI Y,ANDREW J P,RAJIV R S. Process for preparing 1,1,2-trichloro-3,3,3-trifluoropropane:US 8754272B2[P]. 2014-06-17.
[36] MAHLER B A,NAPPA M J. Compositions comprising 2,3-dichloro-1,1,1-trifluoropropane,2-chloro-1,1,1-trifluoropropene,2-chloro-1,1,1,2-tetrafluoropropane or 2,3,3,3-tetrafluoropropene:US8877086B2[P]. 2014-10-04.
[37] WANG H Y,TONG X S. Methods for producing 1-chloro-3,3,3-trifluoropropene from 2-chloro-3,3,3-trifluoropropene:US9000239B2[P]. 2015-04-07.
[38] ZHAI Y,ANDREW J P,RAJIV R S,et al. Stereoselective synthesis of cis-1-chloro-3,3,3-trifluoropropene[J]. Tetrahedron Let.,2016,57(3):396-398.
[39] 王博,张伟,马洋博,等. 第四代制冷剂HFO-1234yf[J]. 化工新型材料,2010,38(8):38-40.WANG Bo,ZHANG Wei,MA Yangbo,et al. The fourth generation refrigerant HFO-1234yf[J]. New Chemical Materials,2010,38(8):38-40.
[40] NAIR H K,POSS A J,NALEWAJEK D,et al. Process for 1-chloro-3,3,3-trifluoropropene from trifluoromethane:US20150045588Al[P]. 2015-02-12.
[41] 韩文锋,靳碧波,周强,等. 三氟甲烷(HFC-23)的资源化转化利用[J]. 化工进展,2014,33(2):483-492.HAN Wenfeng,JIN Bibo,ZHOU Qiang,et al. Conversion and resource utilization of waste CHF3gas[J]. Chemical Industry and Engineering Progress,2014,33(2):483-492.
[42] NAIR H K,DAVID N,POSS A J,et al. Process for 1-chloro-3,3,3-trifluoropropene from trifluoropropene:US9029617B2[P]. 2015-05-12.
[43] 冈本觉,佐久冬彦,项文勤. 反式-1-氯-3,3,3-三氟丙烯的制造方法:103946198A[P]. 2014-07-23.OKAMOTO S,SAKYU F,XIANG W Q. Method for producing trans-1-chloro-3,3,3-trifluoropropene:103946198A[P]. 2014-07-23.
[44] 曹建鹏,温中印,付东海,等. HFO-1234ze在硬质聚氨酯泡沫中的应用研究[J]. 塑料工业,2015,43(9):99-102.CAO Jianpen,WEN Zhongyin,FU Donghail,et al. Application of HFO-1234ze in rigid polyurethane foam[J]. China Plastics Industry,2015,43(9):99-102.
[45] 波克罗夫斯基 K A,默克尔 D C,王 H Y. 联合生产反式-1-氯-3,3,3-三氟丙烯、反式-1,3,3,3-四氟丙烯和1,1,1,3,3-五氟丙烷的集成方法:103189338A[P]. 2013-07-03.POKROVSKI K A,MERKEL D C,WANG H Y. Integrated process to coproduce trans-1-chloro-3,3,3-trifluoropropene,trans-1,3,3,3-tetrafluoropropene,and 1,1,1,3,3-pentafluoropropane:103189338A[P]. 2013-07-03.
[46] 默克尔 D C,约翰逊 R,童雪松,等. 联合生产反式-1-氯-3,3,3-三氟丙烯和反式-l,3,3,3-四氟丙烯的综合方法:102918010B[P].2015-02-25.MERKEL D C,JOHNSON R,TONG X S,et al. Integrated process toco-produce trans-1-chloro-3,3,3-trifluoropropene and trans-1,3,3,3-tetrafluoropropene:102918010B[P]. 2015-02-25.
[47] WISMER J A,BOLMER M S,CHEN B. Azeotrope of HF and 1233zd:US6013846A[P]. 2000-01-11.
[48] 威斯马 J A,陈 B. R-1233与氟化氢的分离:102361842A[P].2012-02-22.WISMER J A,CHEN B. Separation of R-1233 from hydrogen fluoride:102361842A[P]. 2012-02-22.
[49] 冈本正宗,井村英明,高田直门,等. (E)-1-氯-3,3,3-三氟丙烯的制造方法:103930390A[P]. 2014-07-16.OKAMOTO M,IMURA H,TAKADA N,et al. Method for producing(E)-1-chloro-3,3,3-trifluoropropene:103930390A[P]. 2014-07-16.
[50] 科卡利 H,鲍尔 J A,赵 Y,等. 在氟代烃生产中抑制3,3,3-三氟丙炔形成的方法:105228976A[P]. 2016-01-06.KOPKALLI H,BALL J A,ZHAO Y,et al. Process to suppress the formation of 3,3,3-trifluoropropyne in fluorocarbon manufacture:105228976A[P]. 2016-01-06.
[51] YUKIHIRO H,SHUGO H,CHIHIRO S,et al. Measurements of PrT properties,vapor pressures,saturated densities,and critical parameters for R1234ze(Z) and R245fa[J]. Int. J. Refrigeration,2015,52:100-108.
[52] KATSUYUKI T,YUKIHIRO H. PGT property measurements for trans-1,3,3,3-tetrafluoropropene(HFO-1234ze(E)) in the gaseous phase[J]. J. Chem. Eng. Data.,2010,55:5164-5168.
[53] 波克罗夫斯基 K A,辛赫 R R,尚克兰 I,等. 高纯度E-1-氯-3,3,3-三氟丙烯及其制备方法:103402953A[P]. 2012-01-03.POKROVSKI K A,SINGH R R,SHANKLAND I,et al. High purity E-1-chloro-3,3,3-trifluoropropene and methods of making the same:103402953A[P]. 2012-01-03.
[54] 威斯马 J A. 通过萃取蒸馏从氢氯氟烯烃中去除污染物的方法:104144900A[P]. 2014-11-12.WISMER J A. Process for the removal of contaminant from a hydrochlorofluoroolefin by extractive distillation:104144900A[P].2014-11-12.
[55] 冈本正宗,井村英明,高田直门. (E)-1-氯-3,3,3-三氟丙烯的精制方法:104093687A[P]. 2014-10-08.IMURA H,TAKADA N,OKAMOTO M. Method for purifying(E)-1-chloro-3,3,3-trifluoropropene:104093687A[P]. 2014-10-8.
Progress of preparation oftrans-1-chloro-3,3,3-trifluoropropene
ZENG Jijun1,2,HAN Sheng1,2,TANG Xiaobo1,2,ZHAO Bo1,2,HAO Zhijun1,2,ZHANG Wei1,2,LÜ Jian1,2
(1State Key Laboratory of Fluorine & Nitrogen Chemicals,Xi’an 710065,Shaanxi,China;2Xi’an Modern Chemistry Research Institute,Xi’an 710065,Shaanxi,China)
Trans-1-chloro-3,3,3-trifluoropropene [HCFO-1233zd(E)] has virtually zero ozone depletion potential(ODP,0.00024)and very low global warming potential(GWP,7),and therefore it has been proposed as the fourth-generation blowing agent to replace 1,1-dichloro-1-fluoroethane(HCFC-141b)and 1,1,1,3,3-pentafluoropropane(HFC-245fa),due to its very low impact for environment. Synthetic methods of HCFO-1233zd(E) were summarized according to the starting materials. Compared with other routes,the process using HCC-240fa and its derivatives as starting material features good yield and excellent selectivity,and thus has an industrialization prospect. Moreover,the azeotropic separation of HCFO-1233zd(E) with HF and by-product in the preparation process was analyzed and summarized. Developing efficient fluorination catalyst and environmentally friendly purification technology is fundamentally important for industrial synthesis of HCFO-1233zd(E).
trans-1-chloro-3,3,3-trifluoropropene[HCFO-1233zd(E)];blowing agent;synthesis;separation
TQ222.4
A
1000–6613(2017)11–4132–10
10.16085/j.issn.1000-6613.2017-0367
2017-03-07;修改稿日期2017-06-17。
陕西省科技统筹创新工程计划项目(2015KTZDGY05-07)。
曾纪珺(1984—),男,硕士,副研究员,主要从事消耗臭氧层物质替代品的催化合成及其工程化开发。联系人吕剑,研究员,博士生导师,从事氟氮化学品的催化化工领域的研究。E-mail:lujian204@263.net。
