青霉素菌渣堆肥中β-内酰胺酶基因丰度变化
2017-11-07段会英张振华章嫡妮王长永
段会英,赵 娟,张振华,余 冉*,章嫡妮,刘 燕*,王长永
青霉素菌渣堆肥中β-内酰胺酶基因丰度变化
段会英1,赵 娟1,张振华2,余 冉1*,章嫡妮2,刘 燕2*,王长永2
(1.东南大学能源与环境学院,江苏南京 210096;2.环境保护部南京环境科学研究所,江苏南京 210042)
为了解抗生素菌渣堆肥中抗生素残留对抗生素抗性基因(ARGs)环境行为的影响,以青霉素菌渣堆肥为对象,采用实时定量 PCR方法分析了8种典型β-内酰胺酶基因,-TEM、-CTX-M-1、-CTX-M-9、-IMP-1、-VIM-2、-CMY、-OXA-23、-NDM-1在整个堆肥过程中的丰度变化.结果表明,高温堆肥处理大大缩短了青霉素的降解时间;-NDM-1在所有样品中均未检出.通过比较β-内酰胺酶基因在不同处理中第1d和30d的绝对数量变化,在处理组中除-IMP-1、-VIM-2基因绝对数量有所增加外;其他基因都明显减少.从相对丰度看,在堆肥前期,青霉素残留对-TEM、-CTX-M-1、-CTX-M-9、-CMY、-OXA-23、-VIM-2基因有一定诱导富集效应.随着堆肥进程及菌渣堆肥中抗生素浓度的降低,到了堆肥末期,各处理组及对照组-TEM、-CTX-M-1、-CTX-M-9、-CMY的相对丰度较堆肥前期显著降低;而处理组中-IMP-1、-VIM-2的相对丰度较堆肥前期显著增加.
青霉素菌渣;高温堆肥;β-内酰胺酶基因;相对丰度
由于抗生素抗性基因(ARGs)环境污染问题的隐蔽性、滞后性、累积性,及其对人类健康的潜在风险而广受关注.各国学者针对ARGs这种新型环境污染物迅速开展相关基础研究.研究发现[1-2]ARGs在环境中的散播主要以细菌介导的质粒、整合子、转座子、染色体水平转移以及噬菌体转染的方式.而环境介质中的抗生素浓度和活性是诱导ARGs在环境微生物菌群中富集的主要因素.近年来,随着抗生素在医疗和养殖业中的大量使用,导致环境中抗性基因的相对丰度和多样性增加.已有研究表明,在不同环境介质如污水处理厂[1]、畜禽粪便[3]、养殖水域[4]、河流[5]、土壤[6]、冻土[7-8]等中都检测到不同种类和丰度的ARGs.
抗生素菌渣是我国抗生素发酵工业生产中的主要废料,由于其含有抗生素残留及代谢中间产物,于2008年被列入《国家危险废物名录》.其不恰当的处置方式可能会导致残留抗生素进入环境介质中构成潜在的生态风险和资源浪费.前期研究结果发现[9],青霉素菌渣与猪粪的好氧高温堆肥不仅能快速降解青霉素残留,同时青霉素菌渣中大量蛋白和微量元素促进了微生物活性,加速堆肥过程.然而菌渣堆肥中的初始青霉素残留较高,是否会诱导ARGs在微生物群菌中富集,是评价抗生素菌渣堆肥资源化利用技术环境安全风险的主要问题.已有研究表明高温好氧堆肥在一定程度上可以削减禽畜粪便中存在的抗生素耐药菌数量,控制ARGs传播和扩散[10-11].但有关青霉素菌渣堆肥中ARGs的丰度变化研究却鲜有报道.本实验以青霉素菌渣堆肥为对象,基于实时荧光定量PCR(qPCR)技术,研究β-内酰胺酶的8个典型ARGs,-TEM、-CTX-M-1、-CTX-M-9、-IMP-1、-VIM-2、-NDM-1、-CMY、-OXA-23在好氧堆肥不同阶段的丰度变化.初步揭示青霉素菌渣堆肥过程中β-内酰胺酶基因型菌群的变化规律,以期为青霉素菌渣堆肥化的环境安全性评价提供理论依据.
1 材料与方法
1.1 堆肥材料
以青霉素湿菌渣和猪粪为主要原料,以碎木屑为调节剂进行高温好氧堆肥.表1为堆肥原料碳氮比例表(干)及成分比.
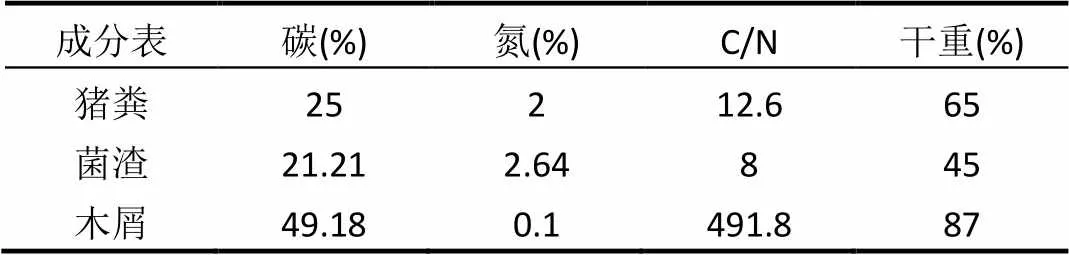
表1 堆肥原料碳氮比例表(干)及成分比
堆肥过程和样品采集方式如赵娟[12]所述,堆制地点选在南京市蔬菜科技园,堆制时间30d.堆肥前期(升温期)人工翻堆1d 1次,之后每隔2d翻堆1次,每次翻堆时间相同.每次翻堆完成后用“5点法”进行样品采集,然后将采集的5个样品混匀,以保证样品的代表性.最终选取第1、6、15、24、30d的样品进行前期处理,用于后续实验分析.表2为所用初始堆肥物料理化性质及组成.
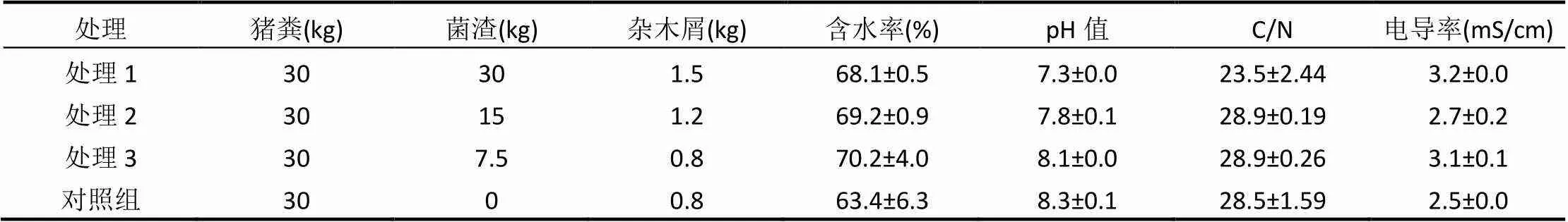
表2 堆肥物料初始理化性质
1.2 青霉素的提取及痕量检测
堆肥样品中青霉素的提取及痕量检测方法:参考马珊珊等[13]方法,经过ASE萃取和SPE净化过程得到的洗脱液过0.22µm尼龙滤膜,然后用外标法及超高效液相色谱方法进行青霉素含量检测.
1.3 堆肥样品DNA的提取
堆肥样品DNA按照FastDNA SPIN Kit for Soil试剂盒说明书进行,用核酸蛋白质分析仪 NanoDrop ND-1000 (NanoDrop Technologies, Wilmington. DE)测定DNA浓度,-20℃保存待测.
1.4 β-内酰胺酶基因及细菌数量的定量PCR (qPCR)的分析
β-内酰胺酶按照Ambler分类法,可分为A、B、C、D 4大类.本次实验从4大类中分别选取了典型的基因分型-TEM、-CTX-M-1、-CTX-M-9、-IMP-1、-VIM-2、-NDM-1、-OXA-23开展其在青霉素菌渣堆肥过程中丰度的变化特征研究. 8种不同基因序列在National Center for Biotechnology Information(简称NCBI)中通过GenBank (http://www.ncbi.nlm.nih.gov/ genbank/) 查找获得合成,具体见表3.本文中所用到引物及探针序列均由生工生物工程(上海)股份有限公司合成.

表3 β-内酰胺酶基因序列
实验中标准曲线的相关系数2值为0.990~1.000,扩增效率范围为95.4%~102%.详细操作方法如下:将合成的质粒进行克隆转化、培养、提取,作为定量PCR的标准模板. β-内酰胺酶基因采用特异性Taqman水解探针法来制作 qPCR 标准曲线.反应用20μL体系,包括Taqman PCR Mix 10μL,上下游引物(= 8000nmol/L)各1.0μL,探针(=4000nmol/L) 0.5μL,超纯水6.5μL,质粒模版1.0μL.同时对细菌16S rRNA基因进行定量,方法是SYBR Green染料法. 20μL扩增体系为SYBR Green Mix 10μL,上下游引物(=100μmol/L)各0.2μL,超纯水8.6μL,质粒模版1.0μL.
将克隆测序得到的标准质粒稀释105~109倍,作为模板构建标准曲线.样品作为定量PCR模板时,稀释10倍,并以无菌水作为阴性对照.所有样品均3个平行,最终计算平均值.表4是实验过程中所用引物和探针以及扩增反应条件.
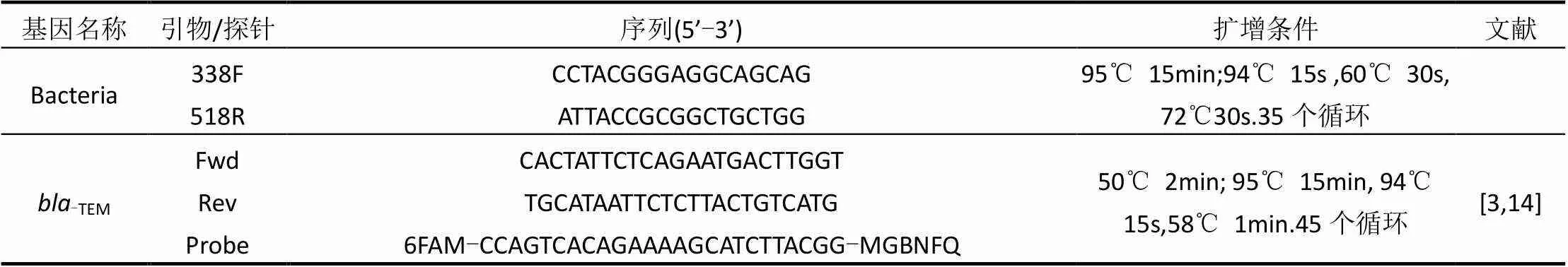
表4 目的基因引物、探针序列及扩增反应条件
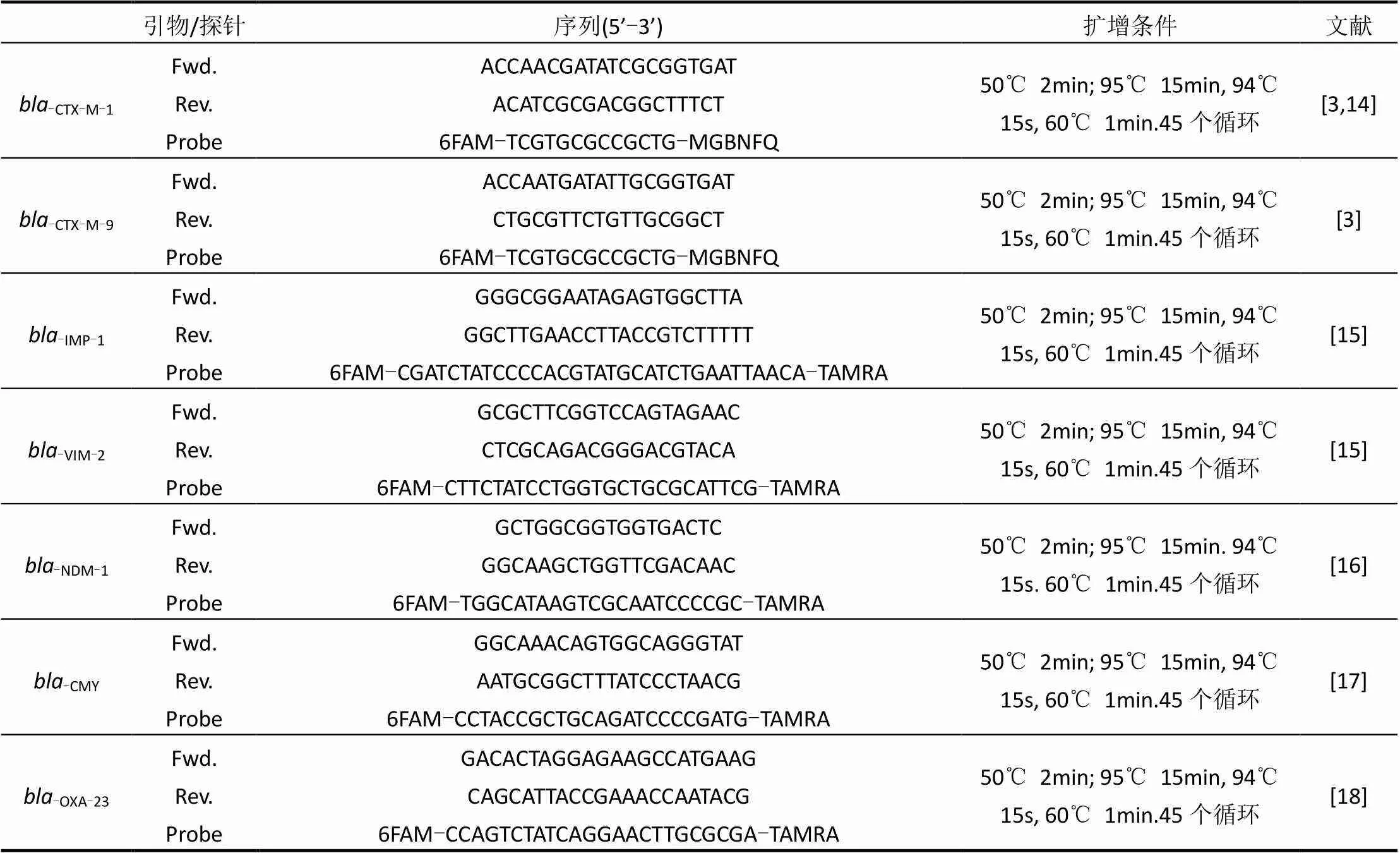
续表4
1.5 数据分析
所得数据用Microsoft Excel 2010进行处理,采用Origin8.5进行作图并用SPSS19.0进行统计学分析.按照公式[19]:相对丰度=β-内酰胺酶基因的拷贝数/16S rRNA基因的拷贝数,计算抗性基因的相对丰度,进行分析讨论.
2 结果讨论
2.1 青霉素残留降解
如图1所示,高温混合堆肥能快速降解堆体中的青霉素残留.处理组1、2、3中青霉素浓度分别从初始的(305.1±16.8)、(208.6±4.2)和(74.9± 3.0)mg/kg降至浓度低于UPLC方法检测限,所用时间分别为9、9、6d.在青霉素菌渣和猪粪的堆肥研究中,有研究称降解99%的青霉素在堆肥前7d内完成[9].青霉素的降解符合一级反应动力学模型[12],处理组1、2、3青霉素钠的降解半衰期(1/2)分别为1.9、2.4、2.3d.
2.2 β-内酰胺酶基因的绝对数量
β-内酰胺酶基因在不同处理中第1d和30d的数量变化如表5所示,堆肥第1d,-TEM、-CTX-M-1、-CTX-M-9、-CMY、-OXA-23、-VIM-2、-IMP-1基因的绝对数量显著高于对照组.一方面,可能由于青霉素菌渣中有机质含量较高且易降解,加速了细菌在堆体内的繁殖数量,同时也促进了携带β-内酰胺酶基因的微生物菌群数量.另一方面,由于青霉素水溶性强,菌渣菌丝体中的青霉素残留快速释放到堆体内,促进了诱导性抗性基因的表达丰度.
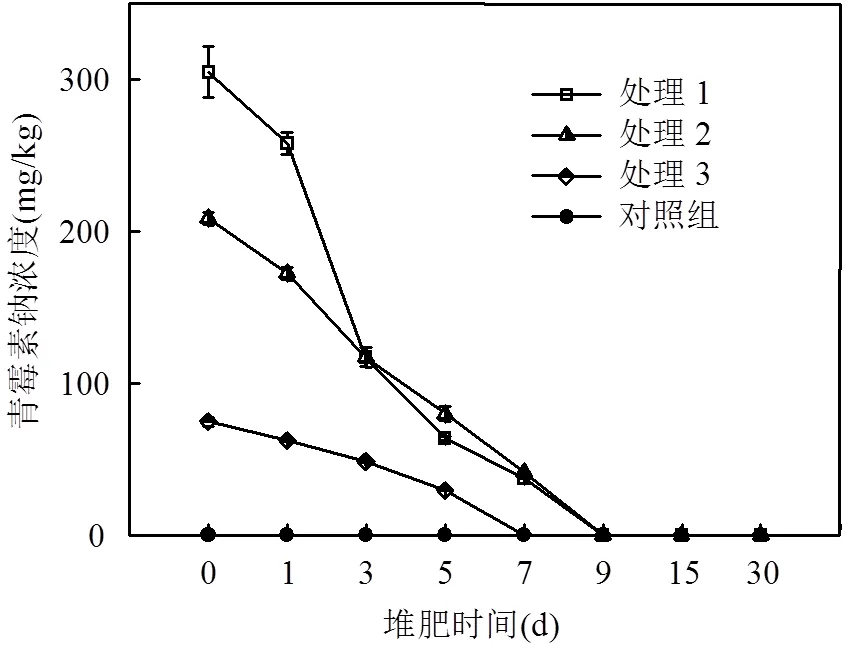
图1 堆肥过程中的青霉素残留浓度

表5 不同处理组中β-内酰胺酶基因在第1d和30d的绝对数量(copies/g DW)
注:#表示同日样品,与对照组存在显著性差异(<0.05):*表示同一处理,与其余天样品存在显著性差异(<0.05).
好氧高温堆肥过程能有效消除β-内酰胺酶基因的数量.处理组中除-IMP-1、-VIM-2基因的绝对数量呈上升趋势,其他基因的绝对数量均显著降低;对照组中的β-内酰胺酶基因除-CMY-OXA-23外均能通过高温好氧堆肥方式得到一定的去除.-NDM-1基因经PCR和qPCR检测结果均显示阴性,表明整个堆肥过程中不存在-NDM-1基因型菌群.
2.3 β-内酰胺酶基因的相对丰度
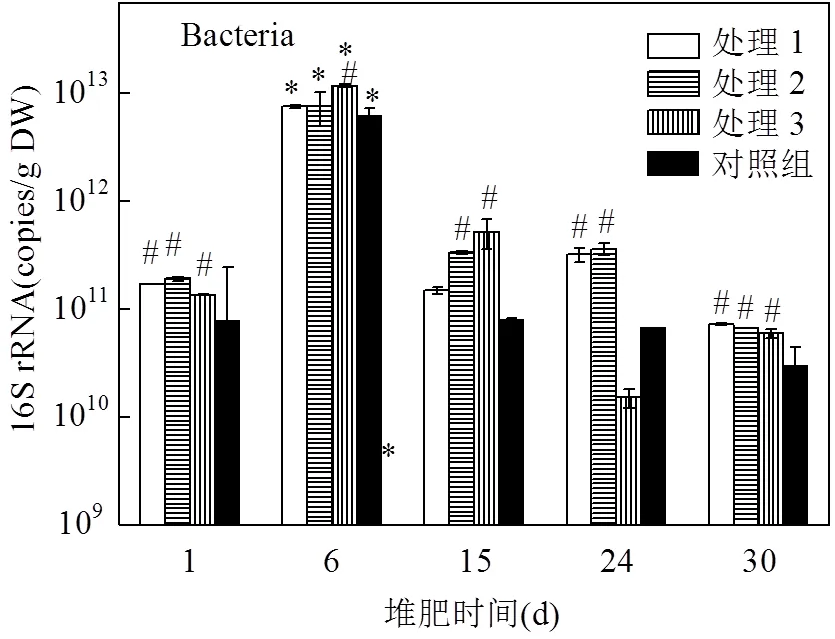
图2 堆肥过程中细菌16S rRNA基因数量变化
#表示同天样品,与对照组存在显著性差异(<0.05):*表示同一处理,与其余天样品存在显著性差异(<0.05)
为了降低堆肥样品DNA提取效率和细菌数量背景值造成的影响.用β-内酰胺酶基因的绝对数量与16S rRNA基因绝对数量的比值(即相对丰度)来分析携带β-内酰胺酶基因型菌群在总细菌群落中所占的平均比例.如图2所示,在堆肥初期和腐熟期处理组的中细菌16S rRNA基因的绝对数量显著高于对照组,说明青霉素菌渣的添加显著提高了堆体中细菌的数量.
2.3.1-TEM基因相对丰度-TEM属于典型的A类超广谱β-内酰胺酶基因(ESBLs),于1965年首次在质粒中发现[20].至2014年已发现了216种亚型[21].主要分布于....和等宿主的质粒中[22].堆肥过程中-TEM基因相对丰度变化如图3所示,在堆肥初期各个处理组中抗性基因-TEM相对丰度在0.4%~3%之间,显著高于其他各个堆肥时期.堆肥高温阶段各处理组的-TEM相对丰度最低.在堆肥腐熟阶段,各个处理组的-TEM相对丰度有所上升(0.05%~0.11%,24d),且无显著差异.堆肥结束后处理1、2、3-TEM基因的相对丰度分别下降了83.6%、42.3%、26.9%.高温堆肥过程虽然在一定程度能消除-TEM基因的相对丰度,但堆肥结束后各处理组的-TEM相对丰度还维持在2×10-3~4×10-3之间.这可能由于-TEM广泛的存在于sp.、sp.、sp.中,而这些菌群又是猪粪堆肥后期的优势菌群[23-24].
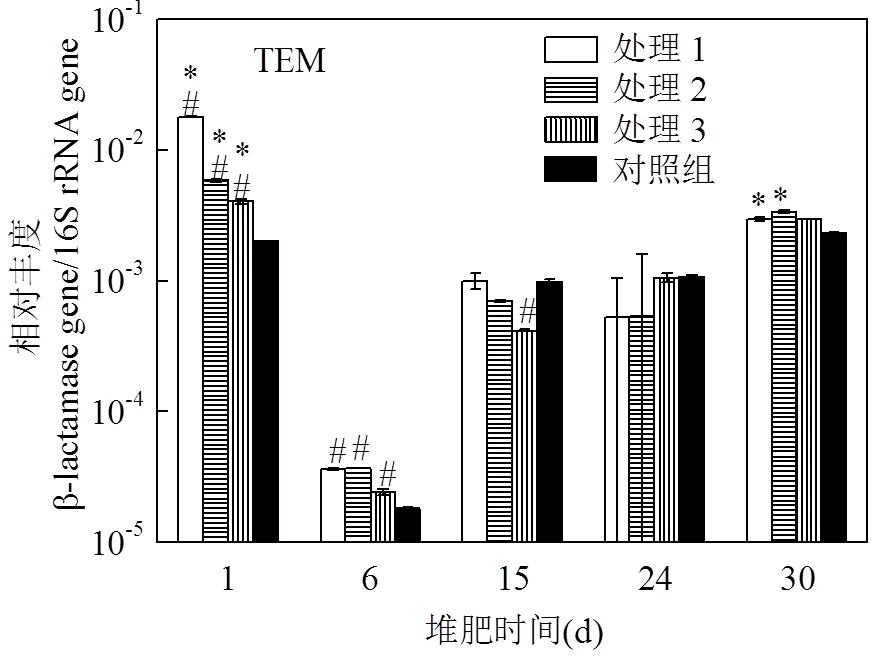
图3 堆肥过程中bla-TEM基因的相对丰度
#表示同天样品,与对照组存在显著性差异(<0.05):*表示同一处理,与其余天样品存在显著性差异(<0.05)
2.3.2-CTX-M基因相对丰度-CTX-M属于A类异源分子结构的β-内酰胺酶基因,目前已发现6种亚型(CTX-M-1、CTX-M-2、CTX-M-8、CTX-M-9、CTX-M-25和KLUC),-CTX-M-1和-CTX-M-9是其主要亚型.-CTX-M最初发现于.sp的染色体上[25],随后研究发现,质粒转染是-CTX-M在不同宿主间转移的主要方式,而禽畜粪便中的.和是其主要携带者[26].大量研究表明,粪便中携带-CTX-M的宿主丰度占到了细菌总丰度的5%~12%[27].如图4所示,不同处理组和对照组中-CTX-M-1和-CTX-M-9基因相对丰度在第1d较高,分别在0.008%~0.1%和0.1%~3%.随着堆肥化过程,-CTX-M基因丰度显著降低.堆肥结束后处理1、2、3和对照组的-CTX-M-1和-CTX-M-9基因相对丰度分别下降了99.7%、96.9%、97.5%、96.1%和99.8%、99.7%、99.8%、99.9%.说明了堆肥化过程能有效消减堆肥原料中的-CTX-M基因丰度.不同处理组及对照组在第1d的-CTX-M基因相对丰度有显著差别,处理1组显著大于其他处理组及对照组,表明在堆肥初期青霉素菌渣中的残留青霉素能够诱导猪粪中的微生物产生青霉素耐药性,进而诱导-CTX-M基因富集.堆肥结束后,不同处理组及对照组的-CTX-M-1基因相对丰度无显著差异,而-CTX-M-9在3个处理组中显著大于对照组.
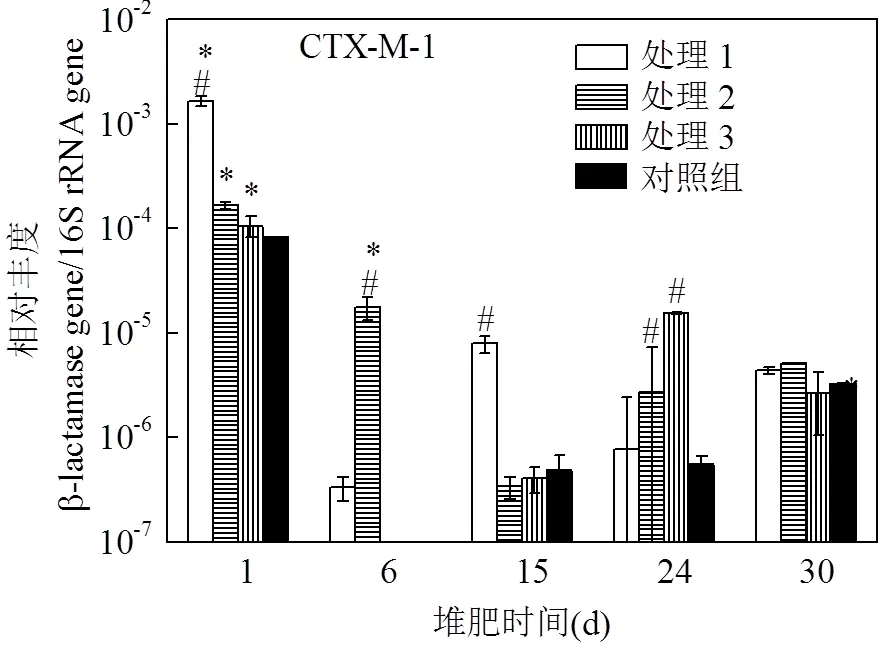
#表示同天样品,与对照组存在显著性差异(<0.05):*表示同一处理,与其余天样品存在显著性差异(<0.05)
2.3.3-IMP-1基因的相对丰度-IMP属于B类金属酶类β-内酰胺酶基因(MBLs),最早在1994年发现于染色体上.至今已发现了42种亚型,其中30多种亚型是在医学临床中的病原菌中发现的[28-29].前期相关研究主要集中于医学临床分离的病原菌中-IMP基因多样性及抗性特征,近年在禽畜粪便堆肥和抗生素广泛使用的近海渔场沉积物中也发现-IMP大量富集.说明了环境介质中的抗生素残留也能诱导-IMP基因在环境微生物中的转移与富集[30-31].如图5所示,堆肥初始1d处理1、2、3中-IMP-1相对丰度分别为4.39×10-6、2.09× 10-6、5.15×10-6显著低于对照组的3.65×10-5.可能是由于堆肥初期-IMP-1主要来源于.sp.,sp.,sp.等热敏性较低的革兰氏阴性病原菌中.青霉素菌渣堆肥较对照组能快速进入高温期,从而有效抑制了-IMP-1宿主的数量.随着堆肥过程,各处理组的-IMP-1基因相对丰度变化的趋势为先减少后增加,至堆肥结束30d,各处理组的-IMP-1相对丰度维持在2×10-5~4×10-4之间.
2.3.4-VIM-2基因的相对丰度-VIM也属于典型的B类金属酶类β-内酰胺酶基因(MBLs),于1997年首次在意大利医院分离的染色体中发现[32].近年来,有研究报道了从饲养猪和家禽农场中分离出携带-VIM基因的和sp.且主要通过IncHI2型质粒在不同宿主中进行基因水平转移[33].如图5所示,青霉素菌渣中的青霉素残留可能对-VIM-2基因在堆肥微生物中的富集产生一定诱导作用.各个堆肥时期,处理1、2、3中-基因相对丰度显著高于对照组.各处理组和对照组中bla-VIM-2基因相对丰度在堆肥前后发生明显变化(>0.05). 堆肥结束后处理1、2、3和对照组分别增加了5.8、2.7、10.4和2.5倍,由于IncHI2型质粒中含有编码抗铜、银、汞、锌等重金属基因,因此禽畜粪便中重金属残留也是诱导-基因在堆肥微生物菌群中富集的机制之一[34].
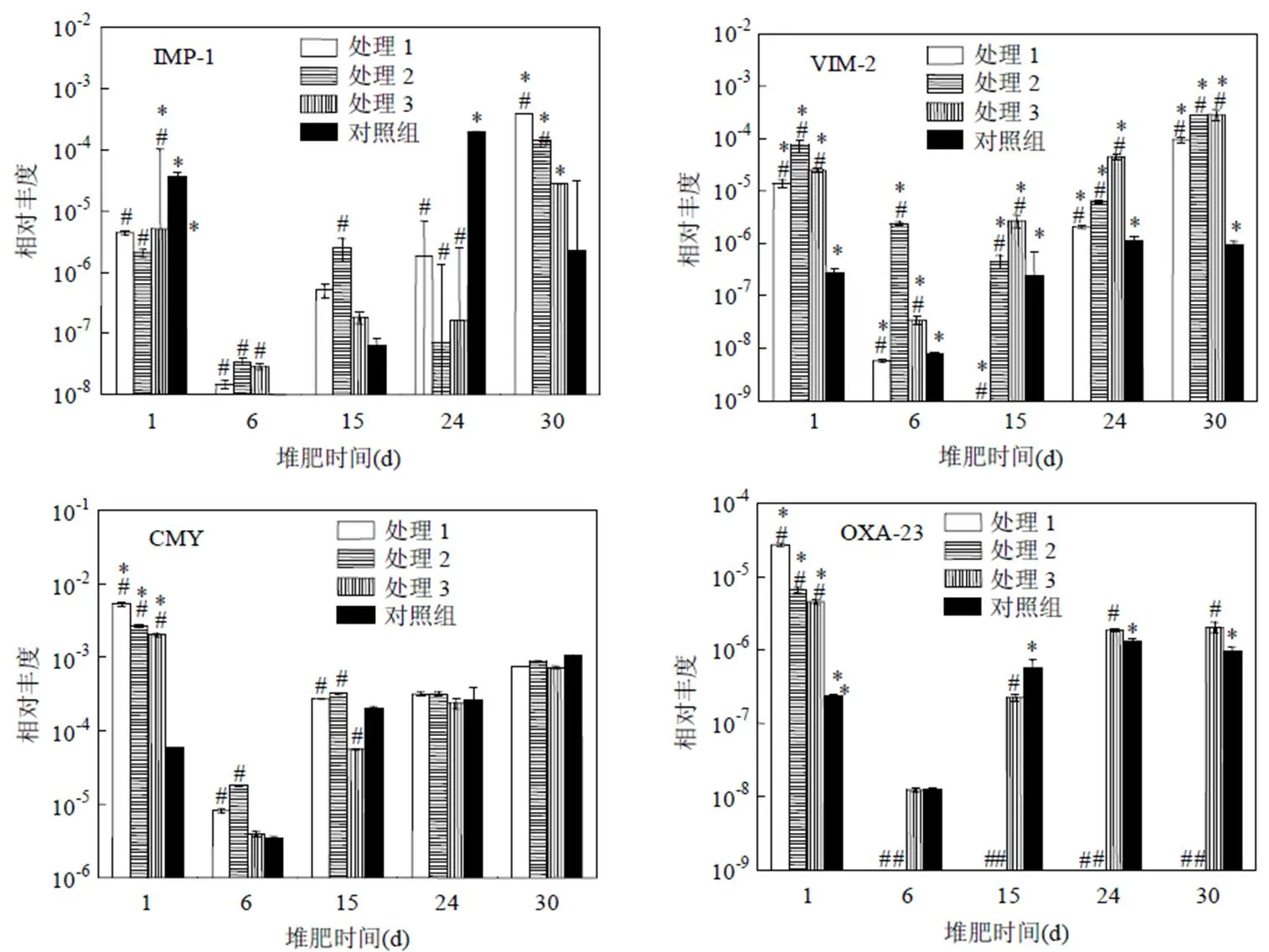
图5 堆肥过程中β-内酰胺酶基因的相对丰度变化
#表示同天样品,与其他组存在显著性差异(<0.05):同一处理,*表示与其他天样品存在显著性差异(<0.05)
2.3.5-CMY基因的相对丰度-CMY属于典型的C类AmpC型β-内酰胺酶基因,最早于1989年在的质粒中发现,至今已经发现43种亚型[35].
近年来,随着抗生素在禽畜养殖业中的广泛使用,从禽畜食品、粪便中分离的、sp.等病原菌中检测出较高丰度的-CMY-2亚型质粒编码基因[36-37].如图5所示,堆肥初始1d处理1、2、3中-CMY相对丰度显著高于对照组.这可能是由于-CMY中部分亚型如-CMY-13、-CMY-11属于底物诱导型,而菌渣堆肥前期中的青霉素残留诱导了相关基因的表达[38].在堆肥高温阶段,各处理组的-CMY基因丰度降到了最低值.随着堆肥进入腐熟期,各处理组的-CMY基因丰度又逐渐上升且无显著差异.至堆肥末期,处理1、2、3中-CMY基因的相对丰度分别下降了86.1%、66.7%、63.5%.
2.3.6-OXA-23基因的相对丰度-OXA属于典型的D类β-内酰胺酶基因,于1993年首次在中发现,目前主要分为4组类群:-OXA-23(OXA-23,-27,-49);-OXA-24(OXA-24,-25,-46,-72);-OXA-58(OXA-58,-96)和-OXA-51,而sp.是其主要的宿主来源[39].如图5所示,处理组和对照组1d中-OXA-23基因的相对丰度分别为2.70×10-5、6.54×10-6、4.52×10-6,显著高于对照组的2.35×10-7.至堆肥结束30d,处理组1、2中-OXA-23基因几乎全部去除,处理3中-OXA-23基因去除55.0%,而对照组增加了3.1倍.
3 结论
3.1 高温堆肥处理大大缩短了青霉素的降解时间,堆肥结束后,处理组中青霉素残留浓度均低于UPLC方法检测限.
3.2 堆肥期间,各处理样品中均未检测到-NDM-1基因.绝对数量上,纯猪粪堆肥对-TEM、-CTX-M-1、-CTX-M-9、-IMP-1有消减作用,青霉素菌渣堆肥对-IMP-1有诱导富集作用对-CMY产生有效削减.
3.3 相对丰度上,在堆肥前期,青霉素残留对-TEM、-CTX-M-1、-CTX-M-9、-OXA-23、-CMY、-VIM-2基因有一定诱导富集效应.至堆肥末期,4个堆体中-TEM、-CTX-M-1、-CTX-M-9、-CMY的相对丰度较堆肥前期显著降低;而处理1、2、3中-IMP-1、-VIM-2的相对丰度较堆肥前期显著增加.
[1] Baquero F, Martínez J L, Cantón R. Antibiotics and antibiotic resistance in water environments [J]. Current Opinion in Biotechnology, 2008,19(3):260-265.
[2] Levy S B, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses [J]. Nature Medicine, 2004,10(12): 122-129.
[3] Colomerlluch M, Imamovic L, Jofre J, et al. Bacteriophages carrying antibiotic resistance genes in fecal waste from cattle, pigs, and poultry [J]. Antimicrobial Agents & Chemotherapy, 2011,55(10):4908-4911.
[4] Hsu J T, Chen C Y, Young C W, et al.Prevalence of sulfonamide- resistant bacteria, resistance genes and integron-associated horizontal gene transfer in natural water bodies and soils adjacent to a swine feedlot in northern Taiwan [J]. J. Hazard Mater., 2014,277(4):34-43.
[5] Li D, Yu T, Zhang Y, et al.Antibiotic resistance characteristics of environmental bacteria from an oxytetracycline production wastewater treatment plant and the receiving river [J].Applied & Environmental Microbiology, 2010,76(11):3444-3451.
[6] Udikovickolic N, Wichmann F, Broderick N A, et al. Bloom of resident antibiotic-resistant bacteria in soil following manure fertilization [J]. Proceedings of the National Academy of Sciences of the United States of America, 2014,111(42):1-6.
[7] D'Costa V M, King C E, Kalan L, et al. Antibiotic resistance is ancient [J]. Nature, 2011,477(7365):457-461.
[8] Lang K S, Anderson J M, Schwarz S, et al.Novel florfenicol and chloramphenicol resistance gene discovered in Alaskan soil by using functional metagenomics [J]. Applied & Environmental Microbiology Aem., 2010,76(15):5321–5326.
[9] Zhang Z, Zhao J, Yu C, et al.Evaluation of aerobic co-composting of penicillin fermentation fungi residue with pig manure on penicillin degradation, microbial population dynamics and composting maturity [J]. Bioresource Technology, 2015,198(8): 403-409.
[10] 田 哲,张 昱,杨 敏.堆肥化处理对畜禽粪便中四环素类抗生素及抗性基因控制的研究进展 [J]. 微生物学通报, 2015, 42(5):936−943.
[11] Wang L, Gutek A, Grewal S, et al. Changes in diversity of cultured bacteria resistant to erythromycin and tetracycline in swine manure during simulated composting and lagoon storage [J]. Letters in Applied Microbiology, 2015,61(3):245-251.
[12] 赵 娟,张振华,段会英,等.青霉素菌渣堆肥过程中青霉素钠降解菌的分离与鉴定 [J]. 环境科学研究, 2016,29(2):271-278.
[13] Ma S, Liu Y, Yu R, et al. Determination of sodium penicillin in soil through accelerated solvent extraction and solid phase extration followed by high performance liquid chromatography [J]. Environmental Chemistry, 2014(11):1978-1985.
[14] Colomerlluch M, Jofre J , Muniesa M. Antibiotic resistance genes in the bacteriophage DNA fraction of environmental samples [J]. PLoS One, 2011,6(3):17549.
[15] Wendel A F, Brodner A H B, Wydra S, et al. Genetic characterization and emergence of the metallo-β-lactamase GIM-1 inspp. and Enterobacteriaceae during a Long-Term outbreak [J]. Antimicrobial Agents & Chemotherapy, 2013,57(10):5162-5165.
[16] Krüttgen A, Razavi S, Imöhl M, et al.Real-time PCR assay and a synthetic positive control for the rapid and sensitive detection of the emerging resistance gene New Delhi Metallo-β-lactamase-1 (bla(NDM-1)) [J]. Medical Microbiology & Immunology, 2011,200(2):137-141.
[17] Geyer C N, Reisbig M D, Hanson N D. Development of a TaqMan multiplex PCR assay for detection of plasmid-mediated amp C β-lactamase genes [J]. J. Clin. Microbiol., 2012,50(11): 3722-3725.
[18] Huang X Z, Cash D M, Chahine M A, et al. Development and validation of a multiplex TaqMan real-time PCR for rapid detection of genes encoding four types of class D carbapenemase in Acinetobacter baumannii [J]. Journal of Medical Microbiology, 2012,61(11):1532-1537.
[19] 任 佳,姚 宏,刘苗苗,等.厌氧和好氧处理过程中四环素抗药基因的丰度 [J]. 中国环境科学, 2016,36(1):268-275.
[20] Datta N, Kontomichalou P. Penicillinase synthesis controlled by infectious R factors in Enterobacteriaceae [J]. Nature, 1965, 208(208):239-241.
[21] 底丽娜,南海辰,夏利宁.TEM型β-内酰胺酶 [J]. 动物医学进展, 2014,35(7):107-110.
[22] Paterson D L, Bonomo R A. Extended-spectrum beta-lactamases: a clinical update [J]. Clinical Microbiology Reviews, 2005, 18(4):657-686.
[23] Zhou S, Nikolausz M, Zhang J, et al. Variation of the microbial community in thermophilic anaerobic digestion of pig manure mixed with different ratios of rice straw [J]. Journal of Bioscience & Bioengineering, 2016,122(3):334-340.
[24] Song C, Li M, Jia X, et al. Comparison of bacterial community structure and dynamics during the thermophilic composting of different types of solid wastes: anaerobic digestion residue, pig manure and chicken manure [J]. Microb. Biotechnol., 2014, 7(5):424–433.
[25] Sarria J C. Infections caused by kluyvera species in humans [J]. Clinical Infectious Diseases, 2001,33(7):69-74.
[26] Zhao W H, Hu Z Q. Epidemiology and genetics of CTX-M extended-spectrum β-lactamases in Gram-negative bacteria [J]. Critical Reviews in Microbiology, 2013,39(1):71-72.
[27] D'Andrea M M, Arena F, Pallecchi L, et al. CTX-M-type β-lactamases: a successful story of antibiotic resistance [J]. International Journal of Medical Microbiology Ijmm, 2013, 303(6/7):305-317.
[28] Osano E, Arakawa Y, Wacharotayankun R, et al. Molecular characterization of an enterobacterial metallo beta-lactamase found in a clinical isolate of Serratia marcescens that shows imipenem resistance [J]. Antimicrobial Agents & Chemotherapy, 1994,38(1):71-78.
[29] Widmann M , Pleiss J.Protein variants form a system of networks: Microdiversity of IMP metallo-beta-lactamases [J]. PLoS One, 2014,9(7):101813.
[30] Zhao Z, Wang J, Han Y, et al.Nutrients, heavy metals and microbial communities co-driven distribution of antibiotic resistance genes in adjacent environment of mariculture [J]. Environmental Pollution, 2016,220:909-918.
[31] Su J Q, Wei B, Ouyang W Y, et al. Antibiotic resistome and its association with bacterial communities during sewage sludge composting [J]. Environmental Science & Technology, 2015,49(12):7356-7363.
[32] Lauretti L, Riccio M L, Mazzariol A, et al. Cloning and characterization of blaVIM, a new integron-borne metallo-beta- lactamase gene from aclinical isolate [J]. Antimicrobial Agents & Chemotherapy, 1999,43(7):1584- 1590.
[33] Fischer J, San J M, Roschanski N, et al. Spread and persistence of VIM-1 Carbapenemase-producing Enterobacteriaceae in three German swine farms in 2011 and 2012 [J]. Veterinary Microbiology, 2016,200:118-123.
[34] Falgenhauer L, Ghosh H, Guerra B, et al. Comparative genome analysis of IncHI2VIM-1carbapenemase-encoding plasmids of Escherichia coli andisolated from a livestock farm in Germany [J]. Veterinary Microbiology, 2015,45(5):609-624.
[35] Jacoby G A. AmpC beta-lactamases [J]. Clinical Microbiology Reviews, 2009,22(1):161-182.
[36] Endimiani A, Hilty M, Perreten V. CMY-2-producingin the nose of pigs [J]. Antimicrobial Agents & Chemotherapy, 2012,56(8):4556-4557.
[37] Mataseje L F, Baudry PJZhanel G G, Morck D W, et al. Comparison of CMY-2plasmids isolated from human, animal, and environmentalandspp. from Canada [J]. Diagnostic Microbiology & Infectious Disease, 2010,67(4):387-391.
[38] Avison M B, Bennett P M, Walsh T R. Beta-lactamase expression in[J]. Journal of Antimicrobial Chemotherapy, 2000,45(6):877-880.
[39] Poirel L, Nordmann P. Carbapenem resistance in: Mechanisms and epidemiology [J]. Clinical Microbiology and Infection, 2006,12(9):826-836.
Abundance dynamics of β-lactamase genes in penicillin biomass-residue composting.
DUAN Hui-ying1, ZHAO Juan1, ZHANG Zhen-hua2, YU Ran1*, ZHANG Di-ni2, LIU Yan2*, WANG Chang-yong2
(1.Department of Energy and Environment Southeast University, Nanjing 210096, China;2.Nanjing Institute of Environmental Sciences, Ministry of Environmental Protection, Nanjing 210042, China)., 2017,37(10):3873~3881
In order to reveal the environmental impacts of antibiotic resistance genes (ARGs) during penicillin biomass-residue composting because of the possible antibiotic residues, the relative abundances and distributions of eight typical β-lactamase genes (-TEM、-CTX-M-1、-CTX-M-9、-IMP-1、-VIM-2、-CMY、-OXA-23、-NDM-1) were investigated with quantitative PCR technique. The results indicated that high temperature composting greatly shortened the degradation time of penicillin. No-NDM-1gene was detected in any sample. The abundances of all studied genes were significantly reduced from day 1 to day 30 in the different penicillin biomass-residue composting experiments except those of the-IMP-1and-VIM-2genes which slight increased. The penicillin residue induced the increases of relative abundance of-TEM,-CTX-M-1.-CTX-M-9,-CMY,-OXA-23, and-VIM-2genes during the early composting stage. With the elongation of the composting process, penicillin residue gradually degraded. At the end of composting, the relative abundances of-TEM、-CTX-M-1、-CTX-M-9、-CMYsignificantly decreased in all treated samples and the control in comparison with those of-IMP-1、-VIM-2, which greatly increased.
Penicillin biomass-residue;composting;β-lactamase genes;relative abundance
X172
A
1000-6923(2017)10-3873-09
段会英(1990-),女,山东临沂人,东南大学能源与环境学院硕士研究生,主要从事污水处理及环境微生物研究.
2017-03-10
国家自然青年科学基金资助项目(41401363,41301278);江苏省自然青年基金资助项目(BK20130102);环境保护部环保公益性行业科研专项(201209024);2016年公益性科研院所基本科研业务费项目
* 责任作者, 余 冉, 副教授, yuran@seu.edu.cn;刘 燕, 副研究员, liuyan@nies.org
