新疆紫草的毛细管电泳指纹图谱研究
2017-11-06张爱芹申刚义
张 蕾 张爱芹 王 嫚 申刚义*
1(中央民族大学 中国少数民族传统医学研究院,北京 100081)2(中央民族大学 生命与环境科学学院 北京市食品环境与健康工程技术研究中心,北京 100081)
新疆紫草的毛细管电泳指纹图谱研究
张 蕾1张爱芹2王 嫚1申刚义*1
1(中央民族大学 中国少数民族传统医学研究院,北京 100081)2(中央民族大学 生命与环境科学学院 北京市食品环境与健康工程技术研究中心,北京 100081)
建立了基于毛细管电泳技术的新疆紫草指纹图谱的质量控制方法。优化后的电泳条件:分离柱为50 μm×40 cm未涂层毛细管柱,运行缓冲液为pH 8.0、100 mmol/L硼酸盐缓冲液(含25 mmol/L SDS及20%(V/V)无水乙醇),进样量0.5 psi×5 s,分离电压25 kV,检测波长214 nm。应用此条件,35 min内可实现新疆紫草有效成分的高效分离。在方法学验证的基础上,建立了新疆紫草指纹图谱。以新疆紫草对照药材指纹图谱为对照图谱,通过特征指纹峰、SF′相似度评价、聚类分析对不同购买地的新疆紫草进行质量评价和鉴别。此研究结果与其它中药鉴定方法对照结果一致。本方法准确、可靠、用时短,且具有良好的重现性,为新疆紫草的质量控制与评价提供了一种新的快速有效的鉴别方法。
指纹图谱; 新疆紫草; 毛细管电泳; 质量控制
2017-07-22收稿;2017-08-29接受
本文系国家自然科学基金项目(Nos. 81573834、81001595)、国家科技支撑计划项目(No.2014BAC15B04)和中央民族大学一流大学一流学科项目(No.YLDX01013)资助
* E-mail: shengy@muc.edu.cn
1 引 言
紫草(Radix Arnebiae)是我国传统中药材,有清热解毒、凉血止痛之功效。紫草类型丰富多样,药性因产地不同而差异明显。2015版《中华人民共和国药典》只收载了新疆紫草(Arnebiaeuchroma(Royle) Johnst)和内蒙紫草(ArnebiaGuttataBunga)两种[1]。其中产自新疆的新疆紫草质量最优,为我国传统名药,有道地药材之誉[2]。新疆紫草的活性成分包括萘醌类紫草素衍生物、生物碱、黄酮类及多糖等。现代医学研究发现,新疆紫草的脂溶性萘醌类活性成分能影响多种肿瘤细胞的增殖和凋亡,是一种很有前景的抗肿瘤特色药材[2,3]。近年来,随着医药行业的科技创新,新疆紫草应用领域拓宽,需求量增大,加之资源日益减少,供需矛盾加大,医药市场出现了以次充好、以假乱真等现象。为保护新疆紫草,我国已将其列为国家二级保护植物。深入开展新疆紫草的质量控制和鉴别研究,对于我国民族地区特色药用资源的可持续发展和开发利用具有重要意义。
2015版中国药典中新疆紫草的鉴别及含量测定主要针对萘醌类成分,包括薄层色谱(TLC)、紫外-可见分光光度法(UV-vis)和高效液相色谱法(HPLC)法。但这些方法评价指标单一,如UV-vis法以羟基萘醌类总色素混合物的含量为质控指标,HPLC法则以总色素单一成分的含量为指标,无法全面反映药材质量特征。近年来,中药指纹图谱作为一种符合中药特色,且国际公认的现代中药及天然药物质量控制模式,逐渐引起研究者的重视[4~8]。特别是基于色谱技术的中药指纹图谱研究最为广泛。如华勇丽等[5]建立了生当归GC-MS指纹图谱。赵春芳等[6]建立了返魂草HPLC指纹图谱。众多学者也先后建立了新疆紫草HPLC指纹图谱[9~11]。研究表明,来自新疆的新疆紫草指纹图谱主要特征大致相同,而非新疆产地的硬紫草及内蒙紫草等与新疆紫草差异明显。目前所建立的新疆紫草HPLC指纹图谱法准确度高、重复性好,但分析时间通常较长(约1 h),且采用单一指纹图谱评价方法,对比性有待提高。相对HPLC运行成本高、分析时间长,色谱柱易污染等问题,毛细管电泳(CE)作为一种高效分离技术,其快速、微量、绿色环保和抗污染能力强等特点,在中药指纹图谱方面有独特优势[12~16]。而迄今尚未见基于CE新疆紫草指纹图谱的研究。本研究在前期相关研究基础上[17,18],以不同购买地、标示为新疆紫草名的药材为鉴别对象,以新疆紫草对照药材为对照品,开展了新疆紫草的CE指纹图谱研究。基于指纹图谱同时运用多种评价体系对样品进行综合质量评价,以期为新疆紫草明辨优劣、去伪存真提供依据。
2 实验部分
2.1仪器与试剂
P/ACE MDQ型毛细管电泳仪(美国BECKMAN COULTER公司),配DAD检测器;未涂层熔融石英毛细管(内径为50 μm,总长为50 cm,有效长度为40 cm,河北永年锐沣色谱器件有限公司);XP205型电子分析天平(瑞士METTLER TOLEDO公司);pH计(SARTORIUS PB-10);KQ-500B型超声波清洗器(昆山市超声仪器有限公司);NW10VF超纯水系统(上海康雷分析仪器有限公司)。
对照品:左旋紫草素(中国食品药品检定研究院);乙酰紫草素(上海源叶生物科技有限公司)。
供试品:新疆紫草对照药材(S1,北京北纳创联生物技术研究院);6种标示为新疆紫草的药材分别购自新疆霍城(S2)、新疆阿克苏(S3)、河北安国(S4)、四川成都荷花池(S5)、新疆伊宁(S6)、云南镇雄(S7);内蒙紫草对照药材(S8,北京北纳创联生物技术研究院)。
β-乙酰氧基异戊酰紫草素和β,β-二甲基丙烯酰紫草素由本实验室前期分离制备获得,结构经NMR和MS鉴定确认。其它化学试剂(国药集团)除特殊说明外均为分析纯;实验用水为超纯水;所用溶液均经过0.22 μm尼龙66膜过滤。
2.2实验方法
2.2.1对照品溶液精密称定0.5 mg左旋紫草素和2.5 mg乙酰紫草素,以运行缓冲溶液定容至10 mL, 得0.05 mg/mL左旋紫草素和0.25 mg/mL乙酰紫草素的对照品溶液,于4℃避光保存备用。
2.2.2供试品溶液参考文献[19],S1~S8粉碎过筛,分别称取2.00 g,每次加入无水乙醇30 mL,超声2次,每次30 min。减压抽滤,将两次滤液合并,旋转蒸发得浓缩液,于4℃避光保存备用,实验前将浓缩液以运行缓冲液定容至20 mL,得0.05 mg/mL供试品溶液。
2.2.3仪器工作条件电泳条件:毛细管柱用前依次用0.1 mol/L NaOH溶液、水、运行缓冲液分别冲洗10 min,进样前用运行缓冲液冲洗5 min。系统控制与数据处理由32Karat软件完成。样品进样量为0.5 psi×5 s,实验温度25℃。

图1 不同类型运行缓冲液中新疆紫草对照药材提取液的电泳图Fig.1 Electrophoregrams of Arnebia euchroma (Royle) Johnst. by capillary electrophoresis in different buffersRunning buffer: a. pH 8.0 and 100 mmol/L NaH2PO4-Na2HPO4+20% (V/V) C2H5OH; b. pH 8.0 and 100 mmol/L Tris-HCl+20% (V/V) C2H5OH; c. pH 8.0 and 100 mmol/L H3BO3-Na2B4O7+20% (V/V) C2H5OH, separation voltage: 25 kV
3 结果与讨论
3.1电泳条件考察
3.1.1检测波长的选择用DAD检测器对新疆紫草对照药材提取液进行波长扫描(200~600 nm)。结果表明,200 nm时,图谱的峰数多但基线噪音大;516 nm的图谱峰数少且信号弱;而214 nm的图谱基线噪音明显降低,峰数多信号强,且重现性增强,利于质量控制,因此最终确定检测波长为214 nm。
3.1.2运行缓冲液的选择新疆紫草主要有效成分为萘醌类化合物,常以左旋紫草素的含量为测定标准。左旋紫草素的pKa=7.59,原则上pH=pKa±1的缓冲液可以作为候选的缓冲液[19]。参照之前的研究[17]发现,增加缓冲液的pH值,可以提高分离效果。本实验选择了pH 8.0的磷酸盐缓冲液、Tris-HCl缓冲液和硼酸盐缓冲液,考察运行缓冲液的种类对紫草提取液分离效果的影响。
由图1可知,磷酸盐缓冲液作为运行缓冲液,体系电导高,基线噪音大,各峰峰形不对称(图1a);Tris-HCl缓冲液作为运行缓冲液,有明显溶剂峰出现,峰信号低(图1b)。硼酸盐缓冲液作为运行缓冲液的电泳图中,峰数多,峰信号明显,组分分离效果最好(图1c)。
实验进一步考察了硼酸盐缓冲液浓度(100~200 mmol/L)的影响。在不同浓度下,电泳图没有明显差别,高浓度硼酸盐会产生更多的焦耳热, 影响分离效果,故选择100 mmol/L硼酸盐缓冲液。
3.1.3有机改性剂的影响萘醌类化合物极性低,当缓冲溶液含有机溶剂时可改善其溶解性,利于分离。且有机溶剂可减小背景电解质的极性,降低Zeta电位,电渗流减小。实验考察了不同体积分数的乙醇(0~30%)对分离的影响。结果表明,当运行缓冲液不含乙醇或含10%乙醇时,各组分分离效果差;当乙醇含量高于10%时,分离效果明显改善;但当乙醇含量高于20%时,分析时间过长。综合分离检测效果,优选含20%乙醇的硼酸盐缓冲液作为运行缓冲溶液,此体系下各组分峰面积及迁移时间重现性良好,基线明显改善。

图2 运行缓冲液中添加SDS前(a)、后(b)新疆紫草对照药材提取液的电泳图Fig.2 Effect of sodium dodecyl sulfonate (SDS) on separation of Arnebia euchroma (Royle) JohnstRunning buffer: a. pH 8.0 and 100 mmol/L H3BO3-Na2B4O7·10H2O +20% (V/V) C2H5OH; b. pH 8.0 and 100 mmol/L H3BO3-Na2B4O7·10 H2O+25 mmol/L SDS+20% (V/V)C2H5OH, separation voltage: 25 kV
3.1.4添加剂的影响表面活性剂不仅对改善和优化CE分离具有重要作用,也可以提高检测的灵敏度。实验考察了常用表面活性剂十二烷基硫酸钠(SDS)对分离的影响。如图2所示,运行缓冲液加入SDS后,各峰信号明显增强,峰面积增大,基线平稳,分离度明显增加且重现性良好。
进一步考察了不同浓度(15~35 mmol/L)的SDS对分离效果的影响。发现随SDS浓度增大,分离度也随之增大,但同时分析时间延长。当SDS浓度大于25 mmol/L时, 1号峰和3号峰迁移时间差值趋于稳定。因此,优选含25 mmol/L SDS的缓冲溶液。
3.1.5分离电压的选择分别以13、16、19、22和25 kV作为分离电压,考察分离电压对分离效果的影响。实验表明,随着电压的增加,柱效明显提高,分离时间缩短且基线保持平稳。在保证分离度的前提下,为缩短分析时间,最终选择25 kV电压。
综上分析,最终确定的CE分离条件为:进样量:0.5 psi×5 s;检测波长:214 nm;分离电压:25 kV;实验温度25℃;pH 8.0、100 mmol/L的含25 mmol/L SDS及20%无水乙醇(V/V)的硼酸盐缓冲液为运行缓冲液。在此条件下,分别对新疆紫草对照药材和内蒙紫草对照药材提取液进行分离。如图3所示,分析时间缩至约30 min, 比HPLC法(>60 min)明显缩短[9,10],且基线和分离度都明显优于已有报道[17,18]。与文献[17,18]相比,样品无需先行预分离制备萘醌类成分,前处理步骤大大简化,可操作性更强。对比图3a和3b还可知,新疆紫草和内蒙紫草的峰数和峰面积都有显著差异。
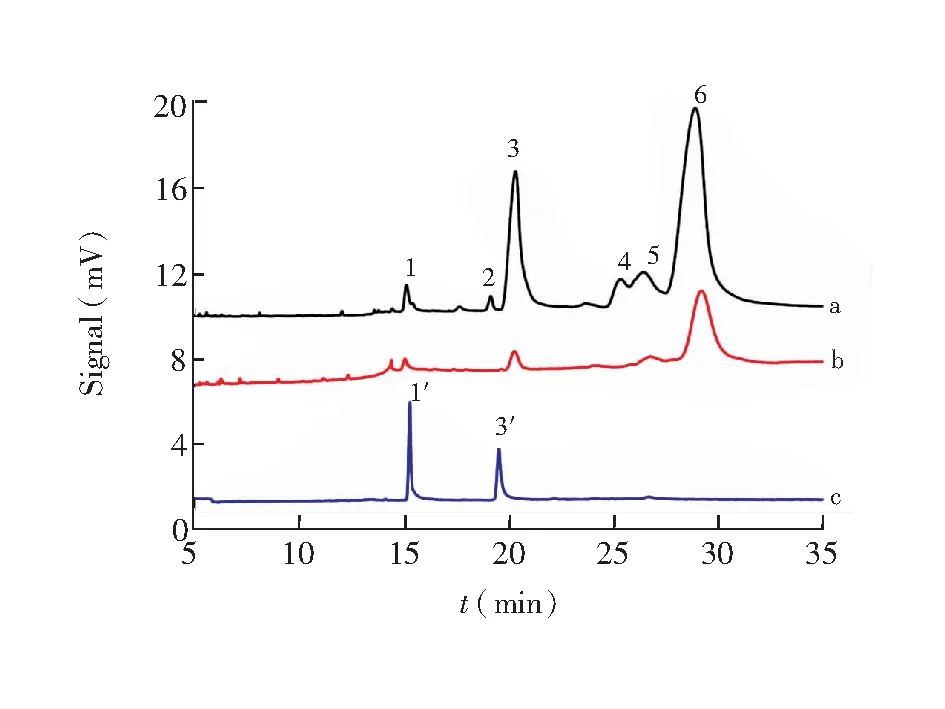
图3 新疆紫草对照药材提取液(a)、内蒙紫草对照药材提取液(b)、左旋紫草素和乙酰紫草素对照品(b)的电泳图;1和1′. 左旋紫草素,3和3′. 乙酰紫草素,4.β-乙酰氧基异戊酰紫草素,6.β,β-二甲基丙烯酰紫草素,2和5.未鉴定成分。
Fig.3 Electrophoregrams of (a)Arnebiaeuchroma(Royle) Johnst., (b)ArnebiaguttataBunge and (c) the mixture of shikonin and acetyl shikonin reference substance solution (c).1 and 1′. Shikonin, 3 and 3′. acetyl shikonin, 4.β-acetoxy isovaleryl shikonin, 6.β,β-dimethylacryl shikonin, 2 and 5:unidentified chemicals.
Running buffer: pH 8.0 and 100 mmol/L H3BO3-Na2B4O7+25 mmol/L SDS+20% (V/V) C2H5OH, separation voltage: 25 kV.
3.2方法学考察
3.2.1专属性实验取左旋紫草素和乙酰紫草素对照品溶液,按上述条件检测,结果见图3C,两组分实现了基线分离,表明本方法具有良好的专属性。采用添加法在新疆紫草对照药材提取液中加入对照品溶液,峰信号增益结果可确定左旋紫草素峰(1号峰)和乙酰紫草素峰(3号峰)。鉴于左旋紫草素与相邻峰分离良好、峰型稳定,且是常见参照物[19],故本实验选左旋紫草素峰作为参照峰。
3.2.2稳定性实验取同一批次的S1药材,按2.2.2节制备供试品溶液,室温密闭保存,分别在0、2、4、8和12 h进行检测,其电泳图共有6个峰,各峰相对保留时间和相对峰面积的RSD分别在1.4%~4.4%、3.1%~5.0%之间,表明供试品在12 h内基本稳定。
3.2.3精密度实验取同一批次的S1药材,按2.2.2节制备供试品溶液,连续进样5次,各峰相对保留时间和相对峰面积的RSD分别在1.0%~3.7%和1.2%~4.3%之间,表明本方法的精密度良好。
3.2.4重现性实验取同一批次的S1药材,按2.2.2节制备供试品溶液,同法制备5个供试品进行检测。各峰相对保留时间和相对峰面积的RSD分别在1.2%~4.7%和1.5%~3.3%之间,表明本方法的重现性良好。
3.3指纹图谱构建与分析
3.3.1指纹图谱建立按2.2.2节制备10批S1供试品溶液,溶液连续进样3次,记录图谱,确定6个出峰稳定且重现性良好的峰为指纹特征峰,见图3a,其为新疆紫草对照图谱。计算列出S1~S8供试品各峰的保留时间(RT)和相对保留时间(以左旋紫草素峰为参照峰,RRT)见表1,峰面积(PA)和相对峰面积(以对照指纹图谱各峰面积为特征指纹向量,RPA)见表2。由表1可知,供试品各峰的RRT的RSD值均小于3%,表明不同供试品中各化学组峰与对照药材重叠性良好。表2则反映出不同来源的新疆紫草各组分的含量差异较大。
表1 不同供试品各峰的保留时间和相对保留时间
Table 1 Retention time (RT) and relative retention time (RRT) of samples from 8 sources

供试品Peak1RT(min)RRT(min)Peak2RT(min)RRT(min)Peak3RT(min)RRT(min)Peak4RT(min)RRT(min)Peak5RT(min)RRT(min)Peak6RT(min)RRT(min)S115.211.0019.191.2620.631.3625.611.6826.641.7529.171.91S215.731.0019.271.2321.351.3626.361.6827.241.7329.321.86S315.321.0019.861.3020.961.3726.721.7427.761.8130.371.98S415.151.0019.001.2520.321.3425.431.6826.291.7428.541.88S515.111.0018.471.2220.651.3725.651.7026.281.7428.841.91S614.771.0019.901.3520.641.4026.631.80--29.441.99S715.341.0019.531.2720.611.3426.061.7027.091.7729.421.92S815.061.00--20.281.35--26.741.7829.201.94RSD(%)-0.00-2.4-1.4-1.7-1.6-2.3注:“-”表示未检出,下同。Note:“-”meansthepeakwasundetectable,Thesameasbelow.
表2 不同供试品各峰的峰面积和相对峰面积
Table 2 Peak area (PA) and the relative peak area (RPA) of samples from 8 sources

供试品Peak1PARPAPeak2PARPAPeak3PARPAPeak4PARPAPeak5PARPAPeak6PARPAS1205291.00110421.002621481.00746661.001440481.008540911.00S2349301.7071100.651339500.51861301.151568811.095015550.59S3218151.0653880.49896860.341108231.481226830.854601280.54S4308581.0651910.472997771.14357770.481609551.124625520.54S5503072.45129711.182450760.94778521.041625841.136475740.76S6266741.30243992.2161580.02208760.28--2574700.30S759750.2941830.38127760.05708080.95642140.45113740.01S878330.38--3054401.17--238120.172540930.30
3.3.2特征指纹峰特征指纹峰的RT及数目能反映样品特有的化学成分及相关性。基于对照指纹图谱并结合表1,计算供试品特征指纹峰的检出数、检出率和重叠率[20]。
由表3可见,除S6(购自新疆伊宁的新疆紫草)特征峰检出率为88.3%,S8(内蒙紫草)的特征峰检出率为66.7%,其余供试品均为100%。特征峰重叠率结果也类似。由此可初步得出S2~S5(新疆霍城、新疆阿克苏、河北安国、四川成都荷花池)和S7(云南镇雄)5种新疆紫草的化学组成与标准品一致。而S6及S8则有较大差异。
表3 不同供试品的特征峰检出率及重叠率
Table 3 Detection of characteristic fingerprint peaks
3.3.3相似度评价特征指纹峰只是简单对指纹图谱定性比较,相似度则通过客观定量描述指纹图谱反映样品的整体特征,是评价样品和标准品一致性程度、控制药材质量的常用方法。
中药指纹图谱相似度评价方法主要有距离系数法、夹角余弦法和相关系数法等。国家药典委员会推荐的《中药色谱指纹图谱相似度评价系统软件2012版》采用夹角余弦法(SF)作为相似度的评价指标,能揭示供试品化学成分与对照品化学成分在分布比例上的相似程度,但无法消除系统中大峰的影响。而在此基础上改进的比率定性相似度SF′,评价时则更为灵敏,对各峰具有等权性,可定性兼定量[21,22]。由图3及表2可知,新疆紫草中各组分的峰面积含量悬殊,差异显著。为消除大峰等因素影
表4 相似度评价
Table 4 Similarity evaluation

供试品SamplesS1S2S3S4S5S6S7S8SF′1.000.920.900.910.910.650.780.64
响,实验采用SF′计算相似度,结果见表4。
相似度值可反映出不同来源药材的品质真假以及优劣。其结果在0.9~1.0符合要求,小于0.8则有显著差异[23]。由表4可知,4个购买地的新疆紫草S2~S5与对照指纹图谱的相似度均≥0.90,表明这4种与对照药材有很好的一致性。结合共有峰率,表明它们的主要化学成分及总体特征与新疆紫草对照药材一致。而S6、S8则差异明显。S7虽然在化学成分上与标准品一致,但相似度差异较大。

图4 样品系统聚类分析图Fig.4 Cluster analysis of samples
3.4系统聚类分析
基于合理的相似度评价并结合数学识别模式,可以相互补充和印证,对中药质控评价更具实际意义[21,24]。在特征指纹峰、相似度评价基础上,本研究进一步运用聚类分析数学识别模式方法,对不同供试品进行品种差异的识别区分。
应用SPSS软件对各供试品图谱的相对峰面积(表2)进行系统聚类分析。采用组间联接法,以夹角余弦为测度,结果见图4。从树状聚类图可以看到聚类趋势,8批供试品分成3类,S1、S2、S4和S5聚为一类,为同一品种; S7和S3品种类似; S6、S8各自成一类。此结果和前两种分析结果相似。
综合特征指纹峰、相似度评价和系统聚类分析,可得出S2~S5(新疆霍城、新疆阿克苏、河北安国和四川成都荷花池)与新疆紫草对照药材品质一致,质量均匀,为新疆紫草; S6(云南镇雄)和S8(内蒙紫草)则差异显著,非新疆紫草; S7(新疆伊宁)和新疆紫草存在差异。
3.5中药鉴定方法学验证
为验证本研究的可靠性和准确性,选取了S2、 S3、 S4、 S6、 S7, 北京市药品检验所中药室对其性状鉴别、显微鉴别和TLC进行质量鉴别。鉴定结果显示S2~S4性状一致,符合我国新疆紫草性状特征; S6与我国新疆紫草性状不符,为进口种; S7与我国新疆紫草性状不符,但同属软紫草属植物,疑为进口种。此验证结果表明本研究建立的新疆紫草CE指纹谱可有效鉴别新疆紫草药材的品质。
4 结 论
本研究建立了新疆紫草CE指纹图谱。在本实验条件下,新疆紫草有效成分分离效率高、分析速度快,所得指纹图谱稳定好、可控性高。通过特征指纹峰、相似度评价和系统聚类分析,对多个购买地的新疆紫草进行了质量评价。实验结果与北京市药品检验所结果一致。本方法可为新疆紫草药材的品质和来源鉴别提供参考。实验结果也初步显示,作为我国四大中药材市场基地的河北安国和成都荷花池药材市场所售的新疆紫草品质好,质量可靠,表明目前我国新疆紫草医药市场发展健康良好。
1ChinesePharmacopoeiaCommission.PharmacopoeiaofThePeople'sRepublicofChina, Part 1. Beijing: The Medicine Science and Technology Press of China,2015: 340
国家药典委员会. 中华人民共和国药典, 一部. 北京: 中国医药科技出版社,2015: 340
2 ZHAN Zhi-Lai, HU Jun, LIU Tan, KANG Li-Ping, NAN Tie-Gui, GUO Lan-Ping .ChinaJournalofChineseMateriaMedica,2015, 40(21): 4127-4135
詹志来, 胡 峻, 刘 谈, 康利平, 南铁贵, 郭兰萍. 中国中药杂志,2015, 40(21): 4127-4135
3 ZHU Meng-Yuan, WANG Ru-Bing, ZHOU Wen, LI Shao-Shun .ActaPharmaceuticaSinica,2012, 47(5): 588-593
朱梦媛, 王汝冰, 周 文, 李绍顺. 药学学报,2012, 47(5): 588-593
4 LI Qiang, DU Si-Miao, ZHANG Zhong-Liang, LYU Chun-Ming, ZHOU Yong-Quan, ZHAO Yan, ZHANG Ning.ChineseTraditionalandHerbalDrugs,2013, 44(22): 3095-3104
李 强, 杜思邈, 张忠亮, 吕春明, 周永全, 赵 燕, 张 宁. 中草药,2013, 44(22): 3095-3104
5 HUA Yong-Li, WEI Yan-Ming, GUO Yan-Sheng, WU Hai-Yan, QU Ya-Ling .ChineseJ.Anal.Chem.,2012, 40(4): 602-607
华永丽, 魏彦明, 郭延生, 吴海燕, 曲亚玲. 分析化学,2012, 40(4): 602-607
6 ZHAO Chun-Fang, LI Qing-Jie, WANG Lian-Ping, HE Zhong-Mei, LIU Yong-Qiang .ChineseJ.Anal.Chem.,2013, 41(1): 133-136
赵春芳, 李庆杰, 王莲萍, 何忠梅, 刘永强. 分析化学,2013, 41(1): 133-136
7 LIU Jie, DING Wen-Jie, HE Bi-Ying, LUO Lan, HUANG Qiang .ChineseJ.Anal.Chem.,2013, 41(4): 500-508
柳 洁, 丁文婕, 何碧英, 罗 兰, 黄 蔷. 分析化学,2013, 41(4): 500-508
8 William P C B, Raquel O R, Juliana J M P.AsianPac.J.TropBiomed.,2017, 7(2): 139-143
9 JIANG Lin, LI Xiao-Jin, JIA Xiao-Guang.ChineseJournalofTraditionalPatentMedicine,2005, 27(11): 1241-1243
姜 林, 李晓谨, 贾晓光. 中成药,2005, 27(11): 1241-1243
10 CAI Ying, LU Yu, GONG Lin, QIU Rong-Li, HUANG Chong-Fa.ChineseJournalofTraditionalMedicalScienceandTechnology,2016, 23(2): 172-174
蔡 鹰, 陆 瑜, 龚 琳, 邱蓉丽, 黄重发. 中国中医药科技,2016, 23(2): 172-174
11 LI Fang-Yue, CAO Xue-Lin, HAO Sheng-Yuan, LIU Hui, XING Shao-Rong, XU Hui.JournalofYantaiUniversity(NaturalScienceandEngineeringEdition),2016, 29(3): 187-192
李方悦, 曹雪霖, 郝盛源, 刘 卉, 邢绍蓉, 许 卉. 烟台大学学报(自然科学与工程版),2016, 29(3): 187-191
12 Jing J F, Mu X Y, Qiao J, Su Y, Qi L.Talanta,2017, 175: 451-456
13 CHEN Yi.CapillaryElectrophoresisandApplications. Beijing: Chemical Industry Press,2006: 6
陈 义. 毛细管电泳技术及应用, 第二版. 北京: 化学工业出版社,2006: 6
14 Mu X Y , Qi L, Qiao J, Yang X Z, Ma H M.Anal.Chim.Acta,2014, 846: 68-74
15 Wang Y, Sun G X, Liu Z B, Liu Y C, Gao Y N, Zhang J Q, Ji Z C, Chen X X .J.Sep.Sci.,2014, 37: 3571-3578
16 XU Jing, WANG Cheng-Fang, DU Shu-Shan, MENG Fan-Yun, LI Feng .ChineseTraditionalPatentMedicine,2014, 36(3): 563-566
许 靖, 王成芳, 杜树山, 孟繁蕴, 李 峰. 中成药,2014, 36(3): 563-566
17 SHEN Gang-Yi, YU Wan-Ting, ZHANG Ai-Qin, LI Shu-Wen, WANG Ning.ChineseJournalofAnalysisLaboratory,2014, 33(5): 513-515
申刚义, 于婉婷, 张爱芹, 李书文, 王 宁. 分析试验室,2014, 33(5): 513-515
18 ZHANG Lei, SHEN Gang-Yi. The 30th Annual Academic Meeting of China Chemical Society,2016, Dalian, China.
张 蕾, 申刚义. 中国化学会第30届学术年会,2016, 大连.
19 LI Quan-Wen, CHEN Zuan-Guang, ZHOU Xie, PAN Ai-Hua, WANG Li-Shi .ChineseJ.Anal.Chem.,2006, 34(7): 991-994
李全文, 陈缵光, 周 勰, 潘爱华, 王立世. 分析化学,2006, 34(7): 991-994
20 CHEN Li-Jing, SUN Xiu-Mei, ZHANG Zhao-Wang, HUANG Yan-Liang, ZHOU Ying .ChineseTraditionalandHerbalDrugs,2012, 43(3): 487-49
陈丽静, 孙秀梅, 张兆旺, 黄延亮, 周 莹. 中草药,2012, 43(3): 487-491
21 SUN Guo-Xiang, HOU Zhi-Fei, ZHANG Chun-Ling, BI Kai-Shun, SUN Yu-Qing .ActaPharmaceuticaSinica,2007, 42(1): 75-80
孙国祥, 侯志飞 , 张春玲, 毕开顺, 孙毓庆. 药学学报,2007, 42(1): 75-80
22 YANG Yan-Tao, WU Chun-Ying, LIU Wen-Long, SHI Ji-Lian, ZHOU Jin, HE Fu-Yuan.ChinaJournalofTraditionalChineseMedicineandPharmacy.2013, 25(8): 1431-1435
杨岩涛, 吴春英, 刘文龙, 石继连, 周 晋, 贺福元. 中华中医药杂志,2013, 25(8): 1431-1435
23 MA Xin, SUN Yu-Qing .ChineseTraditionalandHerbalDrugs,2004, 35(8): 876-878
马 欣, 孙毓庆. 中草药,2004, 35(8): 876-878
24 Dong R F, Su J, Nian H, Shen H, Zhai X, Xin H L, Qin L P, Han T.J.Funct.Foods,2017, 37: 513-522
CapillaryElectrophoresisFingerprintofArnebia
Euchroma(Royle)Johnst.
This work was supported by the National Natural Science Foundation of China (Nos. 81573834, 81001595), the National Science and Technology Support Program, China (No. 2014BAC15B04) and Minzu University of China of the World First-class University and the First-class Discipline (No. YLDX01013).
ZHANG Lei1, ZHANG Ai-Qin2, WANG Man1, SHEN Gang-Yi*1
1(InstituteofChineseMinorityTraditionalMedicine,TheKeyLaboratoryofChineseMinorityTraditionalMedicine
(MinistryofEducationofChina),MinzuUniversityofChina,Beijing, 100081)
2(CollegeofLifeandEnvironmentalScience,BeijingEngineeringResaerchCenterof
FoodEnvironmentandPublicHealthMinzuUniversityofChina,Beijing, 100081)
A method of capillary electrophoresis fingerprint was developed for evaluation of the quality ofArnebiaeuchroma(Royle) Johnst. The samples were separated on a 50 μm×40 cm uncoated capillary separation column at separation voltage of 25 kV with 100 mmol/L borate buffer (pH 8.0) containing 25 mmol/L SDS and 20% (V/V) dehydrated alcohol as running buffer. The injection volume of sample was 0.5 psi×5 s and the detection wavelength was 214 nm. The results indicated that the samples ofArnebiaeuchroma(Royle) Johnst were well separated and detected in 35 min. With Shikonin peak as reference peak, 6 characteristic peaks of standardArnebiaeuchroma(Royle) Johnst were determined. The quality discriminant analyses were accomplished for different kinds of samples namedArnebiaeuchroma(Royle) Johnst that purchased from eight sources by means of characteristic fingerprint peak analysis, similarity evaluation and cluster analysis. This method had good reproducibility, and could be used for the quality control ofArnebiaeuchroma(Royle) Johnst.
Fingerprint;Arnebiaeuchroma(Royle) Johnst; Capillary electrophoresis; Quality control
22 July 2017; accepted 29 August 2017)
10.11895/j.issn.0253-3820.171120
