儿童肝豆状核变性38例临床分析
2017-11-01杨凯华邓朝晖蒋丽蓉
杨凯华 邓朝晖 王 剑 蒋丽蓉
上海交通大学医学院附属上海儿童医学中心(上海 200127)
儿童肝豆状核变性38例临床分析
杨凯华 邓朝晖 王 剑 蒋丽蓉
上海交通大学医学院附属上海儿童医学中心(上海 200127)
目的探讨儿童肝豆状核变性(WD)的临床特征及诊断。方法回顾分析38例WD患儿的临床资料。结果38例患儿中男15例、女23例,确诊时中位年龄6.0岁,起病到确诊时的病程平均5.7个月,中位病程2个月,最长3年。以肝功能异常首发者最为常见(71.1%),其中谷氨酸氨基转移酶>2 倍正常上限者27例(71.1%);血铜蓝蛋白明显降低36例(94.7%),铜氧化酶明显降低37例(97.4%);33例检测24小时尿铜均升高,其中32例(84.2%)>150 μg/24 h。存在K-F环10例(26.3%)。19例患儿行ATP7B基因测序,阳性率83.3%。结论儿童WD起病以肝脏病变为多,结合血铜蓝蛋白、铜氧化酶以及24 h尿铜测定基本可作出临床诊断。对于高度怀疑但依据不足的患儿行ATP7B基因检测有助于早期明确诊断。
肝豆状核变性; 临床特征;ATP7B基因; 儿童
肝豆状核变性又称Wilson disease(WD),发病率约1/10万~1/3万,是一种常染色体隐性遗传性疾病,其相关基因ATP7B定位于13q14.3,编码一种P型铜转运ATP酶,该酶功能异常可导致人体铜代谢障碍,表现为铜经胆汁排泄障碍以及铜与铜蓝蛋白的结合率下降,过量的铜蓄积在肝、脑、肾、角膜等位置,出现相应的脏器功能损害。早期诊断可改善WD患儿的预后,而延误诊断是WD致死致残的主要原因[1]。作为一种遗传代谢性疾病,如能在儿童期甚至婴幼儿期得到明确诊断,可减少致残致死率。
1 临床资料
2010年8月至2016年10月间,上海儿童医学中心共收治并确诊WD患儿38例,其中男23例、女15例,中位年龄6.0岁(3个月~16岁)。
参考Sternlieb标准,具备以下两项及以上者诊断为WD:①具有肝功能损害的临床表现;②有K-F环;③血清铜蓝蛋白降低;④出现震颤、肌肉强直、面具样面容及智力减退等神经系统表现。
38例患儿中,27例患儿因体检肝功能异常就诊,占所有患儿的71.1%;5例以消化道症状起病,分别表现为腹胀、肝脾增大以及消化道出血各1例,黄疸2例;3例以精神神经系统症状起病,分别表现为注意力不集中、步态异常以及言语不清;其余3例中1例以不明原因贫血起病,1例为不明原因血尿,另1例因为姐姐被确诊后来院筛查最终诊断为WD。
38例患儿均进行肝功能、血铜蓝蛋白、铜氧化酶检测。其中肝功能明显异常,谷氨酸氨基转移酶>2倍正常上限(2 ULT)27例(71.1%),结果介于13~594 IU/L,中位数192 IU/L;铜蓝蛋白降低36例(94.7%),正常2例,结果介于0~0.6 g/L,中位数0.07 g/L;铜氧化酶降低37例(97.4%),仅1例正常,结果介于0.01~0.3ΔOD,中位数0.07ΔOD;33例行24小时尿铜测定均升高,其中32例>150 µg/24h,结果介于132 ~3 838 µg/24h,中位数值553 µg/24h。
在获得家属签署知情同意后19例患儿进行基因测序。ATP7B基因变异者18例,其中13例存在明确可致病的复合杂合变异,1例为纯合变异,其余4例仅发现单个变异;1例4岁女性患儿未发现ATP7B基因变异。4例单个变异的患儿临床WD诊断均成立,其中1例3岁男性患儿Sanger测序仅发现单一来自母亲的杂合变异c.3517G>A,p.E1173K,1例16岁男性患儿发现单一的来自母亲的变异c.2333G>T,p.Arg778Leu,另1例是9岁男性患儿,仅发现来自父亲的杂合变异,亦为c.2333G>T,p.Arg778Leu,最后1例5岁男性患儿,仅发现来自父亲的碱基缺失变异c.1803delC,p.Ser602Alafs⋆46。所有变异中,c.2333G>T,p.Arg778Leu最常见 (11/19,57.9%),其余较常见变异为c.2975C>T,p.Pro992Leu(3/19,15.8%)、c.2828G>A,p.Gly943Asp(2/19,10.5%)。见表1。
所有患儿均行头颅MRI检查,5例(13.2%)出现比较典型的基底节、丘脑或豆状核异常信号。
所有患儿均查K-F环,有10例(26.3%)阳性,年龄2~14岁,中位年龄9.5岁,其中1例2岁患儿眼科检查发现角膜周围环形浑浊,考虑K-F环早期表现。
2 讨论
以往研究认为,WD患者大多在20岁左右以肝病起病,或者30岁左右以脑病起病[2]。但近年来小年龄WD患儿多次见诸报道[3-5],甚至有在新生儿中开展筛查的报道[6]。
本组患儿中有27例仅仅是在健康体检时发现肝功能损害,其中24例同时存在24小时尿铜异常和血铜蓝蛋白以及铜氧化酶水平降低进而确诊;其余3例血生化检查支持,因未查24小时尿铜,而是行基因检查,其中2例ATP7B基因检测支持WD诊断,1例发现了单个位点的可致病变异,经过积极治疗肝功能好转。在确诊时2例4岁患儿有非典型的头颅MRI异常,包括双侧脑室后角旁小斑片状异常信号和双侧丘脑异常信号,但其并不存在神经系统异常表现。此27例患儿均获得准确及时的诊断,起病至诊断的平均病程4.5个月,病程中位数2个月。
另外,本组患儿中病程最长的3例,起病至诊断的年龄/病程分别为10岁/2年、10岁/2年和13岁/3年,这3例患儿入院时的症状分别为血尿、口齿不清步态异常以及消化道出血,均已存在肝硬化。其中1例患儿的姐姐年幼时因“溶血,尿毒症”去世,其家长拒绝基因检测,是否同样是WD的肝外表现不得而知;另2例患儿的ATP7B检测结果分别为c.1708-5 T>G(杂合)/c.2333G>T, p.Arg778Leu(杂合)和c.2333G>T,p.Arg778Leu(杂合)/c.2828G>A,p.Gly943Asp(杂合),均提示WD诊断。
在所有38例患儿中,4例表现较特殊。其中1例肝功能或影像学检查无明显变化,该患儿14岁,以头痛、言语不清就诊,头颅MRI存在尾状核豆状核对称性异常信号,K-F环可见,24小时尿铜明显升高且血铜蓝蛋白以及铜氧化酶均明显降低,该患儿未行基因检测。另1例7岁,4岁时发现肝功能异常、24小时尿铜增加以及血铜蓝蛋白降低,在外院曾行ATP7B检测未见有意义变异,3年后出现眼部K-F环,可惜患儿拒绝接受二代测序检测ATP7B基因。第3例患儿血铜蓝蛋白不低,影像学提示肝硬化,低蛋白血症,K-F环可见,基底节异常信号,且ATP7B存在c.2333G>T,p.Arg778Leu(杂合)/ c.2828G>A,p.Gly943Asp(杂合),支持WD诊断。最后1例患儿3月龄开始反复肝功能损害,病初查铜蓝蛋白以及铜氧化酶均正常,3年后查ATP7B提示c.2785A>G,p.929I/c.2333G>T,p.R778L,2处变异分别来自父母,支持WD诊断。
WD的表现非常多样,沿用Sternlieb标准,并不能早期诊断出所有患儿。对于新生儿以及小婴儿,该标准诊断作用有限。首先K-F环以及脑病症状多见于年长儿,本组病例K-F环以及头颅MRI检测阳性率低,分别为26.3%和13.2%;其次铜蓝蛋白的阳性率并非100%,本组中2例铜蓝蛋白不低,但其中1例铜氧化酶降低,本组铜氧化酶降低者达97.4%,略高于铜蓝蛋白;其三,铜蓝蛋白降低的特异性存疑,新生儿尤其是6月龄以内的婴儿,铜蓝蛋白水平普遍明显降低[2,4],若存在其他蛋白质合成或代谢障碍,铜蓝蛋白也会降低;其四,在其他一些慢性肝病患者中同样会出现尿铜水平增加。
作为一种有明确致病基因的单基因遗传病,基因诊断适用于此类患儿。本组2例患儿为亲生姐妹,姐姐确诊WD后,筛查了妹妹和弟弟的ATP7B基因,最终姐妹二人发现同样变异,且妹妹的血生化及24小时尿铜检测支持WD诊断,其弟弟为正常表型携带者。目前基因诊断在不存在其他生化异常仅表现为肝功能异常的小婴儿中的诊断意义已经见诸报道[4]。因此国内外也在探讨新的更敏感的诊断标准,欧美国家在学术会议上相继通过了WD诊断积分,这些新的诊断方案,除了将血、尿等肝、脑外表现囊括在内,同时强调了基因诊断对于诊断WD的重要性[7,8]。
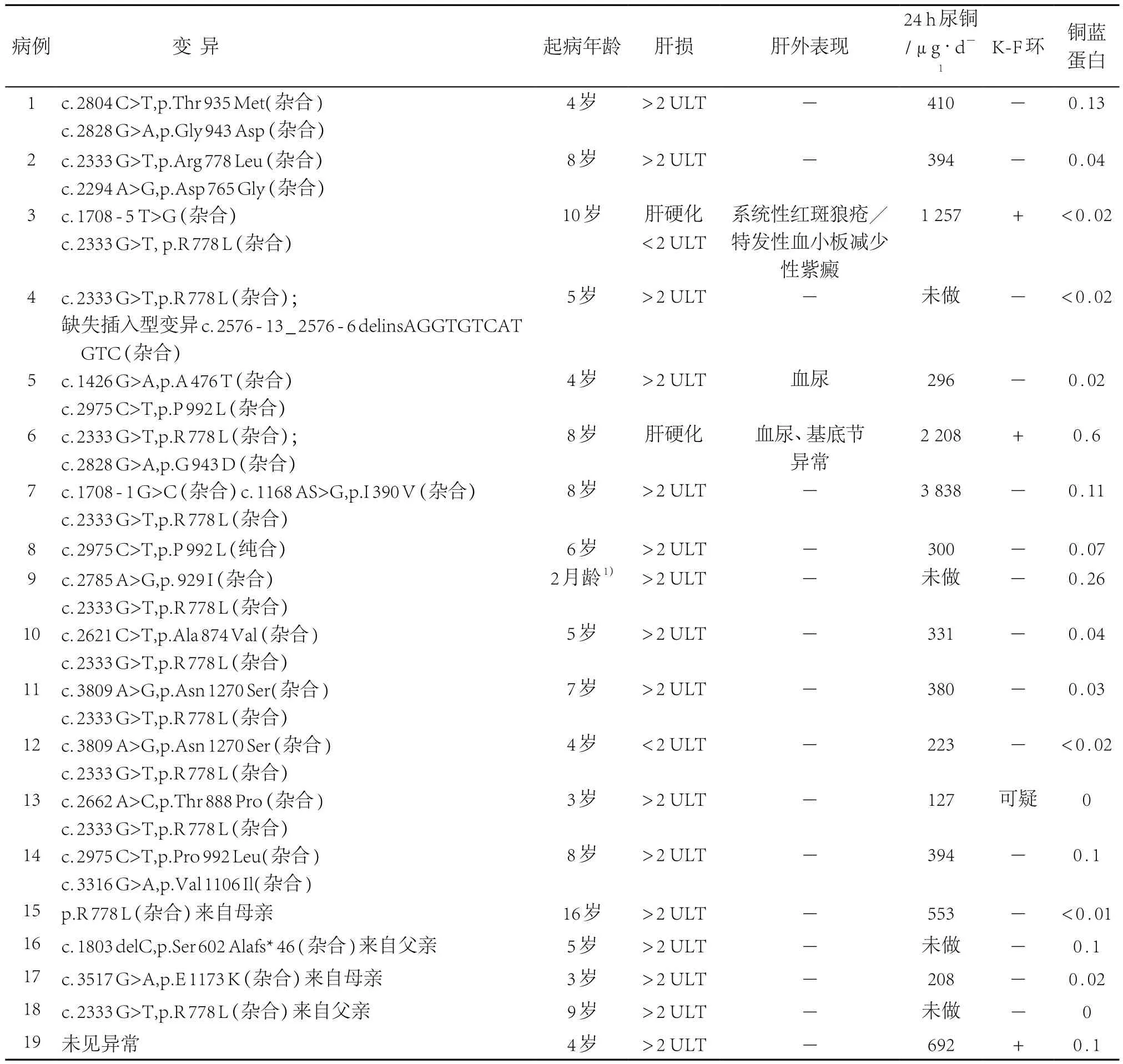
表1 19例行基因测序患儿情况汇总
目前对于WD基因诊断的研究已经不仅仅局限在诊断疾病本身,而是希望能通过早期的基因筛查,可在无症状人群提早进行二级预防,另外通过越来越多的病例推测基因型与表现型的相关性,预测患者可能的临床表现,提高三级预防成功率。目前中国人群中,出现频率最高的变异是p.R778L,这与本组患儿的检测结果相符,本组病例中该变异阳性率高达57.9%,同时也有报道推测该变异与肝脏表现相关[9,10]。本组在2例分别为10岁和13岁存在肝硬化的WD患儿中同样发现了p.R778L变异。此外,2例仅发现单一变异的患儿检测结果均为该高发变异,且出现了症状。似乎提示着该变异与疾病的严重性及起病方式相关,限于目前病例数较少,有待扩大病例数进一步研究。
综上,WD的早期诊断对该病预后意义重大。目前Sternlieb标准可以诊断出绝大多数的WD患儿,但仍有漏诊,且对于婴儿存在过度诊断风险,ATP7B基因检测可提高诊断的准确性。将来对于基因型与表现型相关性的研究,有望提高本病二级以及三级预防的成功率,有较大的社会效益。
[1]Walshe JM.Cause of death in Wilson disease [J].Mov Disord, 2007, 22(15): 216-2220.
[2]Bandmann O, Weiss KH, Kaler SG.Wilson's disease and other neurological copper disorders [J].Lancet Neurol, 2015,14(1):103-113.
[3]Iorio R, D’Ambrosi M, Mazzarella G, et al.Early occurrence of hypertransaminasemia in a 13-month-old child with Wilson disease [J].J Pediatr Gastroenterol Nutr, 2003, 36(5):637-638.
[4]Kim JW, Kim JH, Seo JK, et al.Genetically confirmed Wilson disease in a 9-month old boy with elevations of aminotransferases [J].World J Hepatol, 2013, 5(3): 156-159.
[5]Beyersdorff A, Findeisen A.Morbus Wilson: case report of a two-year-old child as first manifestation [J].Scand J Gastroenterol, 2006, 41(4): 496-497.
[6]Zarrilli F, Elce A, Scorza M, et al.An update on laboratory diagnosis of liver inherited diseases [J].Biomed Res Int,2013, 2013: 697940.
[7]Ferenci P, Caca K, Loudianos G, et al.Diagnosis and phenotypic classification of Wilson's disease [J].Liver Int,2003, 23(3): 139-142.
[8]Roberts EA, Schilsky ML, American Association for Study of Liver Diseases (AASLD).Diagnosis and treatment of Wilson disease: an update [J].Hepatology, 2008, 47(6): 2089-2111.
[9]Xu P, Liang X, Jankovic J, et al.Identification of a high frequency of mutation at exon 8 of theATP7Bgene in a Chinese population with Wilson disease by fluorescent PCR[J].Arch Neurol, 2001, 58(11): 1879-1882.
[10]Liu XQ, Zhang YF, Liu TT, et al.Correlation ofATP7Bgenotype with phenotype in Chinese patients with Wilson disease [J].World J Gastroenterol, 2004, 10(4): 590-593.
Clinical analysis of hepatolenticular degeneration in 38 children
YANG Kaihua, DENG Zhaohui, WANG Jian, JIANG Lirong
(Shanghai Children’s Medical Center Affiliated to Shanghai Jiaotong University School of Medicine, Shanghai 200127, China)
ObjectiveTo explore the clinical characteristics and diagnosis of hepatolenticular degeneration (WD) in children.MethodThe clinical data of 38 children with WD were analyzed retrospectively.ResultsIn the 38 cases (15 males and 23 females), the median age at diagnosis was 6 years, and the average interval between onset and confirmed diagnosis was 5.7 months.The median course of disease was 2 months and the longest was 3 years.Hepatic dysfunction was the most common initial symptom (71.1%), and 27 cases had glutamic acid aminotransferase > 2 ULT (71.1%); Serum ceruloplasmin decreased obviously in 3 cases (94.7%), copper oxidase was significantly reduced in 37 cases (97.4%); 24 h urine copper increased in 33 cases, in which 32 cases (84.2%) had >150 μg/24 h.The K-F rings were presented in 10 cases (26.3%).ATP7Bgene sequencing was performed in 19 cases, and the positive rate was 83.3%.ConclusionsOnset with liver lesions was common in children with WD, The combination of the results of serum ceruloplasmin, copper oxidase, and 24 h urine copper may made a clinical diagnosis.For a highly suspected case with inadequate evidence, theATP7Bgene detected is helpful.
hepatolenticular degeneration; clinical feature;ATP7Bgene; child
doi∶10.3969/j.issn.1000-3606.2017.10.004
蒋丽蓉 电子信箱:jiangl-rong@aliyun.com
2017-03-20)
(本文编辑:蔡虹蔚)
