基于瞬时纳米沉淀法制备尺寸可控载药纳米粒子
2017-11-01,,,,
, , , ,
(华东理工大学化工学院,上海 200237)
基于瞬时纳米沉淀法制备尺寸可控载药纳米粒子
马俊,李莉,王铭纬,周志明,郭旭虹
(华东理工大学化工学院,上海200237)
合成了5种具有不同分子量、不同亲疏水链段比例的两亲性嵌段共聚物——甲氧基聚乙二醇-b-聚己内酯(mPEG-b-PCL),并以其为表面活性剂,采用瞬时纳米沉淀(FlashNanoPrecipitation,FNP)法制备出一系列包裹模型药物β-胡萝卜素的纳米粒子。通过改变两亲性共聚物的结构、分子量、浓度及溶剂体积比(V(H2O)∶V(THF)),成功实现了对纳米粒子尺寸的调控。实验结果表明:聚合物亲水链段分子量比例增大,则纳米粒子尺寸减小;当亲水链段分子量比例相同时,聚合物分子量越大,则纳米粒子尺寸越小;当聚合物质量浓度较高(10.0g/L)时,制备的纳米粒子粒径分布较窄,粒子性能较稳定。
FNP; 聚合物纳米粒子; 尺寸可控
瞬时纳米沉淀 (Flash Nano Precipitation,FNP) 技术由Johnson和Prud’homme在2003年首次提出,是一种可以在极短的时间内制备包含有机活性物质聚合物纳米粒子的新型技术[1]。其流程为:将聚合物、药物溶于有机溶剂(常用四氢呋喃,THF)中,将该股流体与水溶液在CIJ混合器 (Confined impinging jets mixer)中快速对撞混合,从而得到纳米粒子[2]。CIJ混合器工艺流程较简单,且便于操作。但在使用该混合器制备纳米粒子时,要求对撞的两股流体动量必须相等,这极大地限制了研究范围。为了改善不足,Liu等[3-4]进一步完善了CIJ混合器,制备出新型的多通道涡流(MIVM)混合器。与CIJ混合器相比,MIVM混合器采用了4股流体进行混合,且混合方式不再是简单的对撞式混合,而是将流体切向送入混合腔,在混合腔中依靠高剪切力快速进行混合。在这种混合方式下,不仅可以通过调节流速比制备出一系列的纳米粒子,而且经研究证实,与CIJ混合器相比,该混合器可以使混合更有效、更充分。
与传统的透析法[5-11]制备纳米粒子相比,FNP技术的优势在于其大大缩短了纳米粒子的制备时间,整个制备流程在几秒内即可完成。因此,近年来利用FNP技术制备纳米粒子得到了越来越多的关注[12-15]。在利用FNP技术制备纳米粒子过程中,有许多因素如纳米粒子的成核速度、制备过程中反溶剂的用量等都会影响最终形成的纳米粒子的性能[16-17]。Liu等[18]讨论了影响聚合物纳米粒子稳定性的因素,得出了纳米粒子稳定性与聚合物种类以及聚合物药物浓度比有关的结论。Zhu等[19]讨论了嵌段聚合物种类对FNP技术制备纳米粒子稳定性的影响,基于PS-b-PEG、PCL-b-PEG、PLA-b-PEG及PLGA-b-PEG (其中PS、PEG、PCL、PLA、PLGA分别表示聚苯乙烯、聚乙二醇、聚己内酯、聚乳酸、聚乳酸-羟基乙酸)这4种应用广泛的聚合物,利用FNP技术制备包裹β-胡萝卜素的纳米粒子,最终得出了由聚合物PLGA-b-PEG制备的纳米粒子性能较稳定且载药量较高的结论。目前鲜见采用FNP技术制备尺寸可控纳米粒子的文献报道。
在以上文献基础上,本文旨在通过两亲性嵌段共聚物甲氧基聚乙二醇-b-聚己内酯(mPEG-b-PCL)对FNP技术进行进一步的基础研究。在mPEG-b-PCL中,mPEG、 PCL均具有良好的生物相容性,且PCL具有很好的生物降解性,可以被广泛应用于载药领域[20-24]。本文通过调节PEG、PCL两种聚合物的分子量、亲疏水嵌段比例、聚合物浓度以及溶剂体积比(V(H2O)∶V(THF)),利用FNP技术探索实现对载药聚合物纳米粒子尺寸的调控。
1 实验部分
1.1原料及仪器
原料:甲氧基聚乙二醇(mPEG),Mn为5 000、2 000、550,Sigma-Aldrich公司;β-胡萝卜素,纯度97%,Sigma-Aldrich公司;辛酸亚锡,纯度95%,Adamas-beta公司;ε-己内酯,纯度99%,Adamas-beta公司;无水甲苯、乙醚、四氢呋喃、二氯甲烷,分析纯,上海凌峰化学试剂有限公司;高纯氮气,五钢化学气体公司;去离子水,实验室自制。
仪器:电子分析天平,AL104型,梅特勒(上海)仪器公司;磁力搅拌器,DF-101S型,巩义予华仪器有限公司;真空泵,2XZ-2型,临海谭氏真空设备公司;四通道涡流混合器,实验室自制;注射泵,0.01~200 mL/min;电热鼓风干燥箱,DHG-907313S-III型,上海圣科仪器设备有限公司;纳米粒径分析仪,380ZLS型,美国PSS粒度仪公司;氢核磁共振仪,Bruker AM400 MHz型,美国布鲁克(Bruker)公司;凝胶渗透色谱仪(GPC),Waters-1515型,美国Waters公司;荧光分光光度计,F4500型,日本日立公司;透射电镜,JEM-1400型,日本电子股份有限公司。
1.2不同分子量mPEG-b-PCL的合成
mPEG-b-PCL的合成反应式如图1所示。mPEG550-b-PCL3850(下角标分别表示相应物质的分子量,下同)的合成步骤为[25]:将甲氧基聚乙二醇 (mPEG,5.5 g,Mn=550) 溶解在无水甲苯(30 mL)中,随后加入ε-己内酯(PCL,38.5 g,Mn=3 850) 和辛酸亚锡 (150 μL),在氮气保护下,120 ℃搅拌24 h。反应完毕,待冷却至室温后,将产品逐滴缓慢加入大量无水乙醚中。充分搅拌后,抽滤得到白色粉末状固体,转移至真空烘箱中,35 ℃烘干得到最终产物。
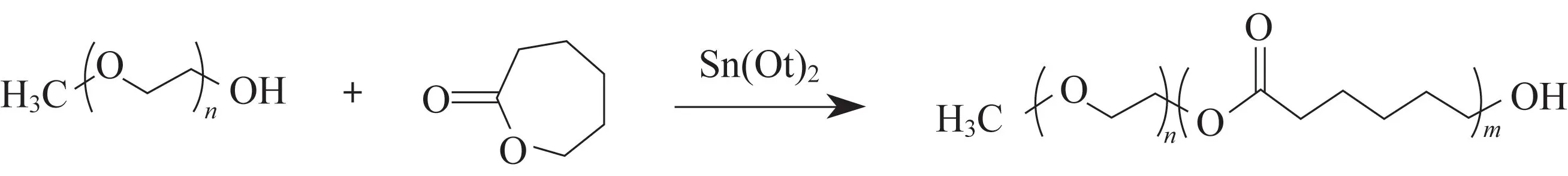
图1 mPEG-b-PCL两嵌段共聚物的合成Fig.1 Synthesis of mPEG-b-PCL di-block copolymer
同上述合成方法一致,采用不同分子量的mPEG和PCL,调节mPEG与PCL的投料比,分别合成出mPEG2000-b-PCL14000、mPEG5000-b-PCL15000、mPEG5000-b-PCL24000和mPEG5000-b-PCL350004种两亲性嵌段共聚物。对最终所合成的产物进行氢核磁表征、GPC表征,并对其临界胶束浓度 (CMC) 进行测定。
1.3用FNP技术制备包裹β-胡萝卜素的纳米粒子
为充分强化组装,实验室自制了一套设备,如图2所示。该设备由2台泵、4支针筒及MIVM混合器组成。其中,泵可以用来控制针筒中的流体流速,可控范围为0~200 mL/min。将β-胡萝卜素以一定比例溶于四氢呋喃(THF)中制备出1号液(Stream 1),聚合物以指定浓度溶于THF中制备出2号液(Stream 2),3号液(Stream 3)和4号液(Stream 4)均为去离子水。随后,设置一定流速,在泵的推动下,让4股流体在混合器中发生瞬间碰撞混合从而得到所需的纳米粒子。
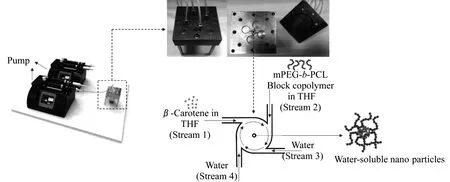
图2 实验流程示意图Fig.2 Experimental flow chart
Russ等[4]已证实,当流体混合为湍流混合时,混合效果更好。实验中通过计算涡流混合器中流体的雷诺数 (Re) 来进行验证。Re的计算公式如下:
(1)
式中:Vi为某一流体的流速,m/s;Qi为某一流体的体积流速,mL/min;Si为流体进口处的通道截面积,本文自制混合器中所有进口处的通道截面积均为1.65×10-6m2;D为混合室的直径,本文中为6×10-3m;υi为流体的运动黏度,水的运动黏度为8.9×10-7m2/s,THF的运动黏度为5.4×10-7m2/s。实验中流速、体积比以及计算得出的雷诺数见表1,其中Re的数值范围为4 500~12 000,均处于湍流混合区,证实所设计的流速符合要求。制备过程中保持时间固定,则水与THF的体积比即为水与THF的流速比。

表1 H2O/THF体积比、流体流速及相应雷诺数
2 结果与讨论
2.1mPEG-b-PCL嵌段聚合物的表征
2.1.1 核磁表征 以mPEG550-b-PCL3850为例,将制备得到的嵌段聚合物配成氘代氯仿溶液进行核磁共振分析,其氢核磁共振图谱如图3所示。化学位移1.38处对应分子式中f处的亚甲基质子峰,2.31处对应d处的亚甲基质子峰,3.38处对应a处的甲基质子峰,3.65处对应b处和c处的亚甲基质子峰,4.06处对应h处的亚甲基质子峰。
mPEG、PCL的实际分子量可由式(2)和式(3)分别计算得出:

(2)
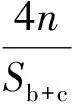
(3)
其中:M为理论分子量;Sb+c表示图3中b+c处的核磁峰面积;Sd表示图3中d处的核磁峰面积;m,n分别表示PCL和PEG中的重复单元数。根据式(2)及式(3)计算可知,产物的实际分子式为mPEG554-b-PCL3899,与设计分子式mPEG550-b-PCL3850接近。由此进一步证明成功合成出产物[25]。

图3 mPEG550-b-PCL3850的氢核磁共振谱图Fig.3 1H-NMR spectra of mPEG550-b-PCL3850
聚合物mPEG2000-b-PCL14000、mPEG5000-b-PCL15000、mPEG5000-b-PCL24000和mPEG5000-b-PCL35000的核磁分析和分子量计算与聚合物mPEG550-b-PCL3850类似,其结果列于表2。
2.1.2 GPC表征 以mPEG550-b-PCL3850为例,使用凝胶渗透色谱仪 (GPC) 测定其分子量以及分子量分布 (PDI) 。测试流动相为四氢呋喃,标定物为聚苯乙烯。测试结果如图4所示。由此得到样品mPEG550-b-PCL3850实际分子量为4 440,分子量分布为1.44。从图4可以看出,分子检出越早,分子量越大,符合GPC检测的一般规律。

图4 嵌段聚合物GPC数据Fig.4 GPC data of block copolymers
2.1.3 临界胶束浓度(CMC)的测定 采用芘荧光探针法测试所合成的嵌段聚合物的CMC值[26]。具体测试步骤如下:将6 mg聚合物溶解于1 mL四氢呋喃中,剧烈搅拌下,缓慢加入30 mL蒸馏水,形成内相为有机相,外相为连续水相的乳剂,聚合物重排形成胶束。将乳剂敞口搅拌,除去挥发有机相,配成质量浓度为0.2 g/L的聚合物胶束母液。与此同时,称取1 mg芘溶于1 L去离子水中,连续搅拌3 d制得芘的饱和溶液。取10个5 mL棕色直形罗口瓶,用芘的饱和水溶液对聚合物母液进行稀释,分别稀释成0.1,0.01,5×10-3,2.5×10-3,1.0×10-3,5×10-4,2.5×10-4,1.0×10-4,1.0×10-5g/L和1.0×10-6g/L共10个质量浓度不同的溶液,之后使用振荡器连续振荡5 min并静置数小时,使聚合物充分自组装。配样完成后,转移2 mL样品于比色皿中,置于仪器内测试。
以mPEG550-b-PCL3850为例,用荧光光度计的氙灯作为激光光源,激发波长固定为335 nm,狭缝宽度为3 mm,波长扫描范围350~450 nm。由于芘在372 nm和383 nm处的荧光强度的比值 (I372/I383) 强烈依赖于溶剂的极性,故当芘增溶于聚合物形成的胶束后,芘所处环境极性的变化会引发I372/I383突变,由此可确定聚合物的CMC值。芘在不同浓度聚合物溶液中的荧光光谱图如图5(a)所示,计算I372/I383,将其作为y轴,将lgρ(ρ为嵌段聚合物质量浓度)作为x轴,进行作图,最终可通过拐点处切线的交点计算得出聚合物CMC值,如图5(b)所示。
采用相同方法可得到其余各分子量聚合物的CMC值,结果列于表2中。分析数据可知,各分子量聚合物的CMC值符合非离子表面活性剂CMC值的一般变化规律,即疏水端链段越长,CMC值越小;聚氧乙烯聚合度越高,CMC值越大。这也间接证明了合成的聚合物分子量符合设计值,同时为聚合物表面活性剂自组装的浓度取值提供参考。
mPEG-b-PCL嵌段聚合物核磁表征、GPC表征以及CMC测试最终结果如表2所示,其中,fPEG为实际合成的嵌段聚合物中亲水链段所占的分子量比例。由此可知,成功合成出不同分子量的嵌段聚合物。
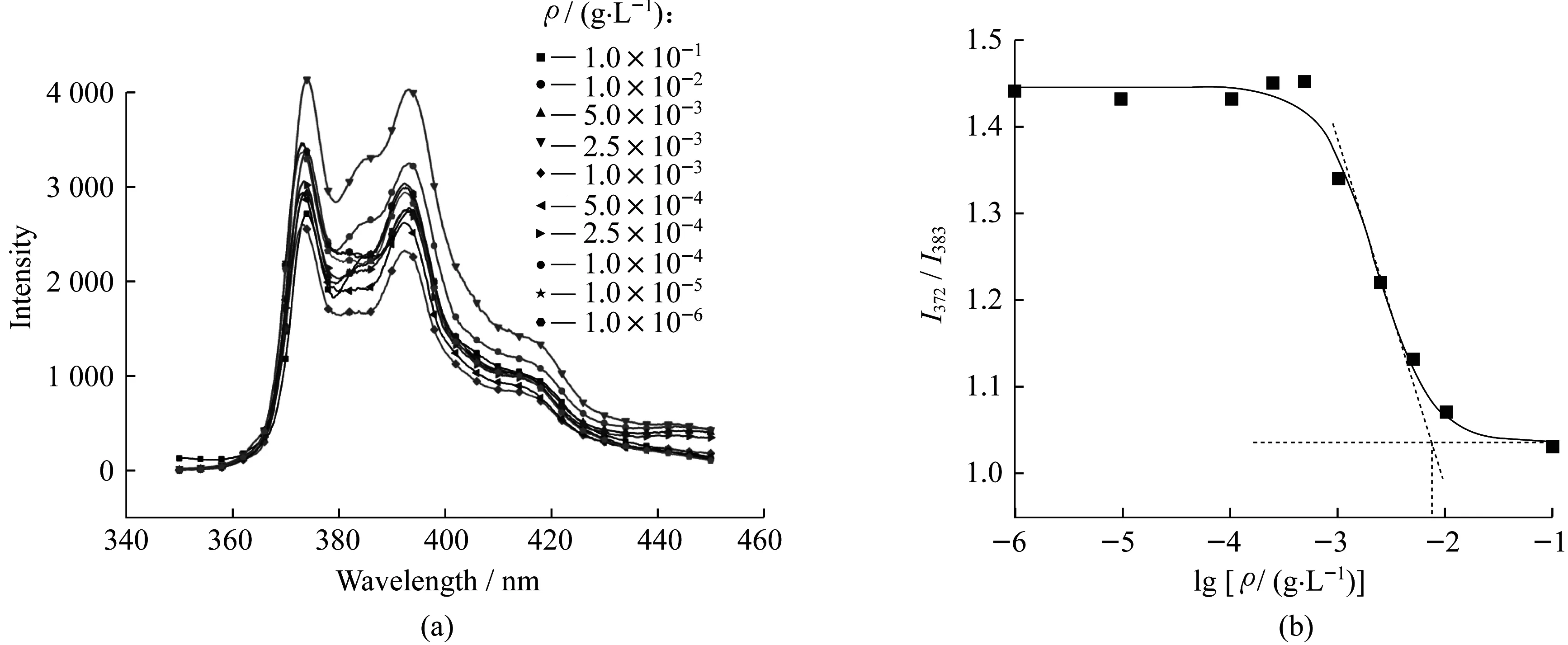
图5 芘在不同浓度聚合物(mPEG550-b-PCL3850)溶液中的荧光光谱(a);mPEG550-b-PCL3850CMC值拟合图(b) Fig.5 Fluorescence spectra of pyrene in polymer mPEG550-b-PCL3850 solution with different concentrations (a) and CMC fitting chart of mPEG550-b-PCL3850 (b)

表2 mPEG-b-PCL两亲性嵌段共聚物核磁表征、GPC表征及CMC测试结果
a)Tested by 1H-NMR;b)Tested by GPC;fPEG—MmPEG/MmPEG-b-PCL
2.2聚合物纳米粒子包封率、载药率的测定
2.2.1β-胡萝卜素标准曲线的绘制 配制水与THF体积比为4∶1的溶液A。称取一定质量的β-胡萝卜素溶于A中,配制一系列浓度的β-胡萝卜素的A溶液,将其分别置于紫外分光光度计中测试,结果如图6所示。
由图6可知,溶液A在460 nm处有最大吸光度,取460 nm处吸光度对质量浓度作线性回归,如图7所示,得到回归方程为y=0.075x+0.05,R2=0.976,可见在所选质量浓度范围内,β-胡萝卜素的质量浓度与紫外吸收度线性关系良好。
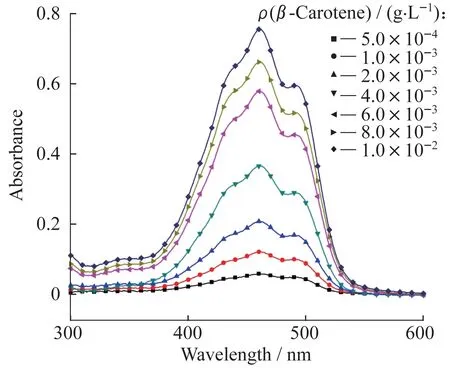
图6 不同浓度下β-胡萝卜素紫外光谱图Fig.6 UV absorbance spectra of β-carotene in different concentrations
2.2.2 载药纳米粒子包封率、载药率的计算 取5 mL制备的纳米粒子溶液(初始溶液),先透析除去其中的THF,此时会有部分未被包裹的β-胡萝卜素沉降出来,用450 nm的滤头过滤掉沉降出的β-胡萝卜素。将滤液冻干,12 h后取出,将其重新分散至5 mL溶液A中得到溶液B,以溶液A为对照,在460 nm处测定溶液B中β-胡萝卜素的紫外吸收值,代入标准曲线即得被包裹的β-胡萝卜素的浓度,从而求出胡萝卜素包裹量,根据下式算出纳米粒子包封率及载药率。
包封率=β-胡萝卜素包裹量/β-胡萝卜素给药量×100%
摩尔载药率=β-胡萝卜素被包裹物质的量/(β-胡萝卜素给药物质的量+聚合物物质的量)×100%
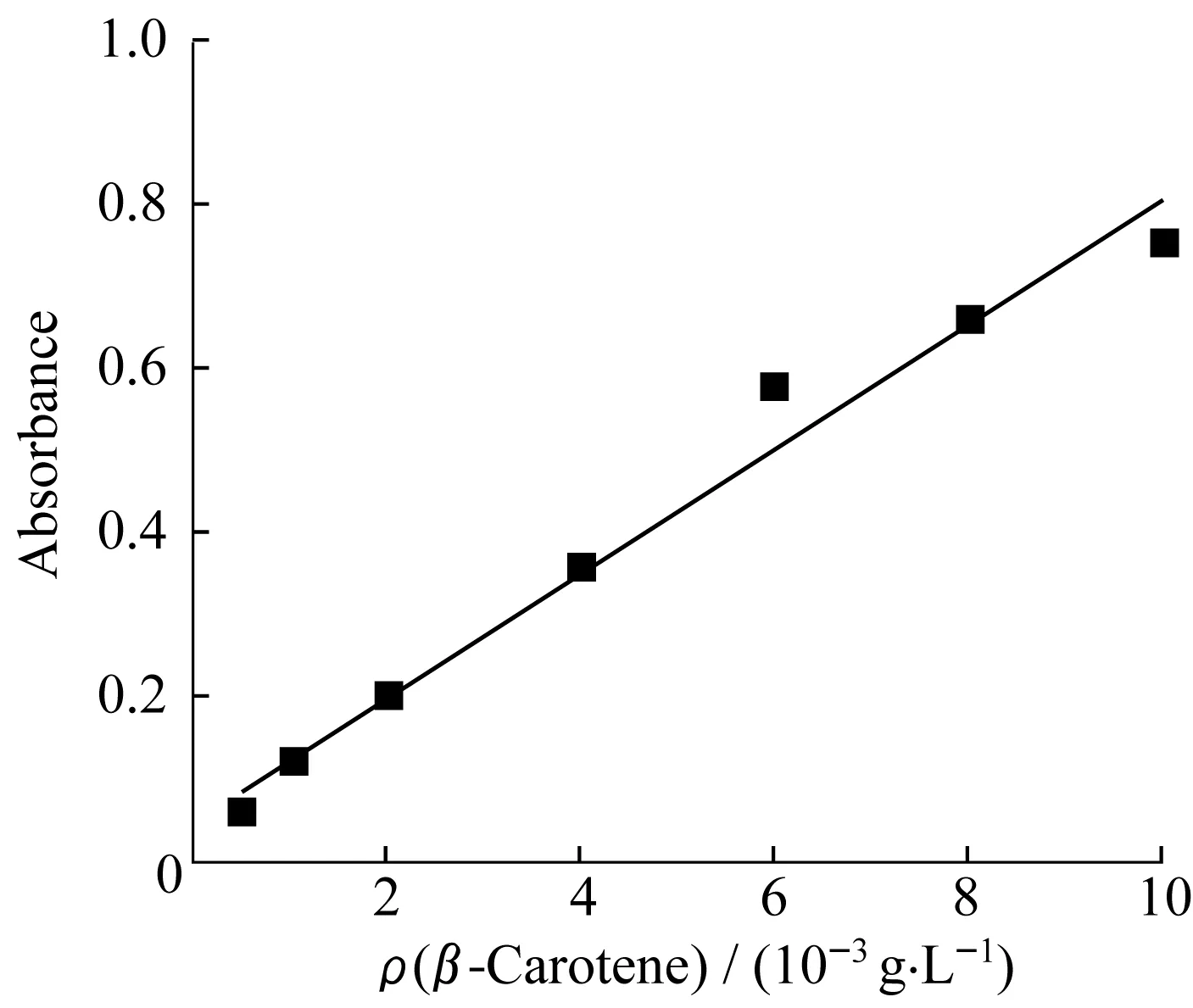
图7 β-胡萝卜素在溶液A中的紫外标准曲线Fig.7 UV standard curve of β-carotene in solution A
根据实验结果计算得出,由聚合物mPEG550-b-PCL3850、mPEG2000-b-PCL14000、mPEG5000-b-PCL15000、mPEG5000-b-PCL24000和mPEG5000-b-PCL35000制备得到的纳米粒子的包封率分别为35.3%、73.6%、77.4%、71.5%和82.1%,可见分子量较小的聚合物mPEG550-b-PCL3850的药物包封率明显低于其他样品,包封效果较差,随后章节将对其粒子结构作进一步的表征;纳米粒子的摩尔载药率分别为3.3%、18.0%、22.4%、27.9%、38.0%,误差范围内仍然是分子量较小的聚合物mPEG550-b-PCL3850摩尔载药率最低,可见载药率对聚合物的分子量有最低阈值要求。
2.3采用FNP技术制备并调控纳米粒子的尺寸
2.3.1 聚合物浓度及溶剂体积比对纳米粒子尺寸的影响 以mPEG5000-b-PCL35000为例,采用DLS测试了聚合物在不同浓度及溶剂体积比条件下形成的纳米粒子的尺寸及其分布,实验结果如图8所示。当聚合物质量浓度分别为0.1、1.0、10.0 g/L时,纳米粒子PDI分别在0.3、0.2和0.1附近。这表明,随着聚合物质量浓度的增加,纳米粒子分散性越来越窄。这可能是由于流体在混合腔内进行瞬间混合时,聚合物浓度过低会导致机械混合作用力过大,抵消了聚合物疏水缔合的能力,使得形成的纳米粒子大小非常不均匀,分散性差。因此,在使用FNP技术制备纳米粒子过程中,聚合物浓度需要达到一定的数值。为减小实验误差,降低粒子分散性,将后续实验中的聚合物质量浓度固定为10.0 g/L。

图8 溶剂体积比不同时纳米粒子的分散性 Fig.8 PDI of nano particles in different solvent ratios
同时,随着V(H2O)∶V(THF)的增加,纳米粒子粒径逐渐减小的趋势非常明显。当V(H2O)∶V(THF)由4∶1增加到13∶1时,纳米粒子尺寸由210 nm减小至130 nm,降低了近40%,如图9所示。分析原因,可能是由于随着溶剂体积比的增加(水相比例增加),β-胡萝卜素与水相之间的极性排斥作用逐渐增强,从而使得纳米粒子结构更加紧凑,导致纳米粒子尺寸减小,可见β-胡萝卜素的成核诱导作用不容忽视。
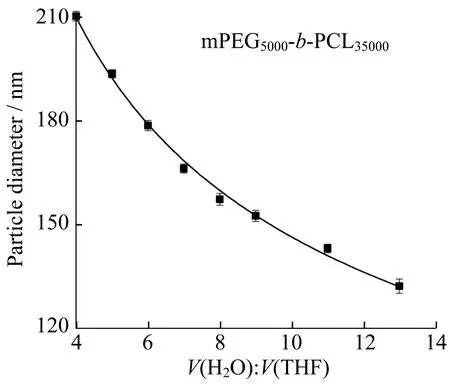
图9 溶剂体积比不同时的纳米粒子粒径Fig.9 Particle size of nano particles in different solvent ratios
此外,本文还研究了纳米粒子的稳定性。将在较高质量浓度(10.0 g/L)下制备得到的mPEG5000-b-PCL35000纳米粒子静置数天(5 d,10 d),利用DLS追踪其粒径变化,结果如图10所示。从图中可以看出,所制备出的纳米粒子较稳定,粒径大小并无显著变化。用其他不同分子量聚合物制备得到的纳米粒子粒径与分散性变化规律与mPEG5000-b-PCL35000相似。
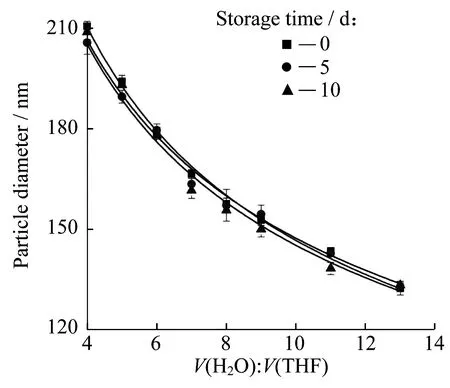
图10 mPEG5000-b-PCL35000纳米粒子粒径大小与储存时间的变化关系
综上,当聚合物质量浓度较高(10.0 g/L)时,采用FNP技术制备的纳米粒子分散性较好,且粒径随着溶剂体积比V(H2O)∶V(THF)的增加而减小。由此可见,调节聚合物质量浓度及溶剂体积比是控制聚合物纳米粒子尺寸行之有效的手段。
2.3.2 聚合物亲水链段分子量比例对纳米粒子尺寸的影响 在较高质量浓度(10.0 g/L)下,实验设计了mPEG5000-b-PCL15000(fPEG=24.99%) 、mPEG5000-b-PCL24000(fPEG=17.23%)和mPEG5000-b-PCL35000(fPEG=12.50%) 3种不同亲水链段分子量比例的聚合物,采用FNP技术制备一系列纳米粒子,利用DLS测试其粒径,结果如图11所示。

图11 聚合物亲水链段分子量比例对粒子尺寸的影响Fig.11 Effect of hydrophilic segment ratio on particle size
从图中可以看出,随着聚合物亲水链段分子量比例的增加,纳米粒子粒径逐渐减小。与前面溶剂体积比的作用类似,这可能同样是由于聚合物分子中亲水链段增大,使得β-胡萝卜素与水相之间的极性排斥作用逐渐增强,从而使纳米粒子结构更加紧凑,导致纳米粒子尺寸减小。
综上,聚合物亲水链段分子量比例对最终形成的纳米粒子粒径有较大的影响,通过改变聚合物亲水链段分子量比例,可以实现对纳米粒子粒径的调控。
2.3.3 聚合物分子量对纳米粒子尺寸的影响 在相同质量浓度(10.0 g/L)和相同亲水链段分子量比例(fPEG=12.50%)下,聚合物分子量对纳米粒子粒径的影响如图12所示。实验设计了mPEG550-b-PCL3850、mPEG2000-b-PCL14000和mPEG5000-b-PCL350003种分子量不同的嵌段聚合物,采用FNP技术制备一系列纳米粒子,利用DLS测试其粒径。

图12 聚合物分子量对粒子尺寸的影响Fig.12 Effect of molecular weight on particle size
从图12中可以看出,由3种不同分子量嵌段聚合物制备得到的纳米粒子尺寸从大到小依次为mPEG550-b-PCL3850、mPEG2000-b-PCL14000、mPEG5000-b-PCL35000,即随着分子量的增加,纳米粒子尺寸逐渐减小。这个结果比较出乎预期,分析原因可能是当聚合物分子量较低时,聚合物分子链的体积排斥效应不强,使得更多的聚合物分子容易聚集成为一个纳米粒子,即每个聚集体是由更大物质的量的聚合物组装而成的,粒径也就越大。
同时,由于聚合物分子量较小,形成每个聚集体的聚合物的物质的量也就很难在短时间内得到均匀控制。根据图12所示PDI数据可知,聚合物分子量越小,形成的纳米粒子分散性就越大。对比2.2.2节聚合物的载药率和药物包封率数据可知,纳米粒子并非尺寸越大载药效率越高,聚合物胶束的组装紧密度才是决定载药率的重要因素。
综上,聚合物分子量对纳米粒子尺寸及分散性影响较大,当分子量较大时,纳米粒子分散性较好且尺寸较小。
2.3.4 聚合物纳米粒子TEM表征 利用透射电镜对mPEG550-b-PCL3850、mPEG5000-b-PCL15000及mPEG5000-b-PCL35000制备得到的纳米粒子形貌进行表征,结果如图13所示。可见所制备得到的纳米粒子形态呈较均一的球形,并且从左到右粒径逐渐减小,符合前文所述规律,即聚合物亲水链段分子量比例越大,所制得的纳米粒子粒径越小;当聚合物亲水链段分子量所占比例相同时,聚合物分子量越大,所制得纳米粒子的粒径越小。
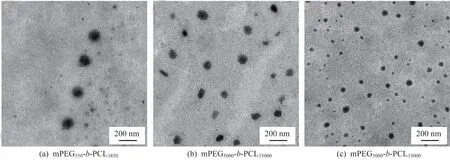
图13 纳米粒子TEM示意图Fig.13 TEM image of nano particles
3 结 论
(1) 聚合物浓度及溶剂体积比显著影响用FNP技术制备的纳米粒子尺寸及分散性。在较高质量浓度下,得到的粒子分散性较好;且随着溶剂体积比V(H2O)∶V(THF)的增大,粒子的尺寸逐渐减小。
(2) 聚合物中亲水链段分子量比例对纳米粒子尺寸影响较大。聚合物亲水链段分子量比例越大,纳米粒子粒径越小,分散越均匀。
(3) 聚合物分子量对纳米粒子尺寸、分散性和载药率有一定的影响。当聚合物浓度及聚合物亲水链段分子量比例相同时,聚合物分子量越大,所制得的纳米粒子尺寸越小,分散越均匀。聚合物的载药率和药物包封率对分子量也有一定要求,分子量太小不利于载药。
[1] JOHNSON B K,PRUD′HOMME R K.Mechanism for rapid self-assembly of block copolymer nanoparticles[J].Physical Review Letters,2003,91(11):1-4.
[2] JOHNSON B K,PRUD′HOMME R K.Chemical processing and micromixing in confined impinging jets[J].AIChE Journal,2003,49(9):2264-2282.
[3] LIU Ying,CHEN Chungyin.Mixing in a multi-inlet vortex mixer (MIVM) for flash nano-precipitation[J].Chemical Engineering Science,2008,63(11):2829-2842.
[4] RUSS B,LIU Ying,PRUD′HOMME R K.Optimized descriptive model for micromixing in a vortex miner [J].Chemical Engineering Communications,2010,197(8):1068-1075.
[5] BLANAZS A,ARMES S P,RYAN A J.Self-assembled block copolymer aggregates:From micelles to vesicles and their biological applications[J].Macromolecular Rapid Communications,2009,30(4/5):267-277.
[6] SUN Huanli,GUO Bingnan,LI Xiaoqing,etal.Shell-sheddable micelles based on dextran-SS-poly(ε-caprolactone) diblock copolymer for efficient intracellular release of doxorubicin[J].Biomacromolecules,2010,11(4):848-854.
[7] ZHULINA E B,ADAM M,LARUE I,etal.Diblock copolymer micelles in a dilute solution[J].Macromolecules,2005,38(12):5330-5351.
[8] AATHIMANIKANDAN S V,SAVARIAR E N,THAYUMANAVAN S.Temperature-sensitive dendritic micelles[J].Journal of American Chemical Society,2005,127(42):14922-14929.
[9] JI Yongqiang,WANG Weishan,LI Ganzuo,etal.Rheological properties of wormlike micelles formed in the sodium oleate/trisodium phosphate aqueous solution[J].Chinese Chemical Letters,2008,19(4):483-487.
[10] MILLER T,COLEN G Van,SANDER B,etal.Drug loading of polymeric micelles[J].Pharmaceutical Research,2013,30(2):584-595.
[11] MATSUOKA K,TAKAGI K,HONDA C.Micelle formation of sodiumhyodeoxycholate[J].Chemistry and Physics of Lipids,2013,172/173(3):6-13.
[12] GINDY M E,PRUD′HOMME R K.Multifunctional nanoparticles for imaging,delivery and targeting in cancer therapy[J].Expert Opinion Drug Delivey,2009,6(8):865-878.
[13] WANG Mingwei,XU Yisheng,WANNG Jie,etal.Biocompatible nanoparticle based on dextran-b-poly(L-lactide) block copolymer formed by flash nanoprecipitation [J].Chemistry Letters,2015,44(12):1688-1690.
[14] WANG Mingwei,YANG Nan,GUO Zhiqian,etal.Facile preparation of AIE-active fluorescent nanoparticles through flash nanoprecipitation[J].Industrial & Engineering Chemistry Research,2015,54(17):4683-4688.
[15] AKBULUT M,GINART P,GINDY M E,etal.Generic method of preparing multifunctional fluorescent nanoparticles using flash nano precipitation[J].Advanced Functional Materials,2009,19(5):718-725.
[16] GINDY M E,PANAGIOTOPOULOS A Z,PRUD′HOMME R K.Composite block copolymer stabilized nanoparticles:Simultaneous encapsulation of organic actives and inorganic nanostructures[J].Langmuir,2008,24(1):83-90.
[17] ADDIO D,PRUD′HOMME R K.Controlling drug nanoparticle formation by rapid precipitation[J].Advanced Drug Delivery Reviews,2011,63(6):417-426.
[18] LIU Ying,TONG Zhen,PRUD′HOMME R K.Stabilized polymeric nanoparticles for controlled and efficient release of bifenthrin[J].Pest Management Science,2008,64(8):808-812.
[19] ZHU Zhengxi.Effects of amphiphilic diblock copolymer on drug nanoparticle formation and stability[J].Biomaterials,2013,34(38):10238-10248.
[20] GOU Maling,ZHENG Xiuling.Self-assembled hydrophobic honokiol loaded mPEG-PCL diblock copolymer micelles[J].Pharmaceutical Research,2009,26(9):2164-2173.
[21] SHEN Yang,LENG Mengtian,YU Hongchi,etal.Effect of amphiphilic PCL-PEG nano-micelles on HepG2 cell migration[J].Macromolecular Bioscience,2015,15(3):372-384.
[22] PENG Wei,JIANG Xinyi,ZHU Yuan,etal.Oral delivery of capsaicin using mPEG-PCL nanoparticles[J].Acta Pharmacologica Sinica,2015,36(1):139-148.
[23] WANG Qin,JIANG Jiayu,CHEN Wenfei,etal.Targeted delivery of low-dose dexamethasone using PCL-PEG micelles for effective treatment of rheumatoid arthritis[J].Journal of Controlled Release,2016,230:64-72.
[24] LINCE F,MARCHISIO D L,BARRESI A A.Strategies to control the particle size distribution of poly-ε-caprolactone nanoparticles for pharmaceutical applications[J].Journal of Colloid and Interface Science,2008,322(2):505-515.
[25] HWANG Minji,SUH Ju myung,BAE You han,etal.Caprolactonic poloxamer analog:PEG-PCL-PEG[J].Biomacromolecules,2005,6(2):885-890.
[26] SHANNIGRAHI M,BAGCHI S.Novel fluorescent probe as aggregation predictor and micro-polarity reporter for micelles and mixed micelles[J].Spectrochimica Acta:Part A.Molecular and Biomolecular Spectroscopy,2005,61(9):2131-2138.
PreparationofSize-ControllableDrugLoadedNanoParticlesBasedonFlashNanoPrecipitationMethod
MAJun,LILi,WANGMing-wei,ZHOUZhi-ming,GUOXu-hong
(SchoolofChemicalEngineering,EastChinaUniversityofScienceandTechnology,Shanghai200237,China)
Five amphiphilic diblock copolymers,methoxy poly(ethylene glycol)-b-polycaprolactone (mPEG-b-PCL) with different molecular weights and hydrophilic/hydrophobic proportions were synthesized.A series of nano particles encapsulated model drug beta-carotene and protected by the prepared copolymers as surfactants were prepared by using flash nano-precipitation (FNP) method.The size and size distribution of nano particles were controllable by changing the block structure,molecular weight,polymer concentration and solvent ratio (V(H2O)∶V(THF)).Experimental results showed that the size of nano particles decreased by increasing the solvent ratio or hydrophilic proportion.Under the same proportion of the hydrophilic moieties,the size of particles was decreased by increasing the molecular weight.The nano particles became more stable and their size distribution was narrower upon increasing polymer concentrations.
FNP; polymeric nano particles; size-controllable
TQ317
A
1006-3080(2017)05-0597-09
10.14135/j.cnki.1006-3080.2017.05.001
2017-01-10
国家自然科学基金(21476143)
马 俊(1994-),女,硕士生,主要从事高分子表面活性剂的合成及聚集态结构研究。
李 莉,E-mail:lili76131@ecust.edu.cn
