血清生物标志物在中国儿童X-连锁慢性肉芽肿病早期诊断的META分析
2017-11-01陈少华应艳琴
陈少华,应艳琴
(华中科技大学同济医学院附属同济医院 儿科,湖北 武汉430030)
血清生物标志物在中国儿童X-连锁慢性肉芽肿病早期诊断的META分析
陈少华,应艳琴*
(华中科技大学同济医学院附属同济医院 儿科,湖北 武汉430030)
目的探讨中国儿童X-连锁慢性肉芽肿病(X-CGD)临床特点及早期血清生物标志物特征,为早期确诊提供线索。方法文献检索2017年6月前明确报道关于中国X-CGD患儿临床表现和血清生物学数据的文献,并进行meta分析和总结,寻找早期血清学特征。结果检索具备上述特点的已发表文献包括病例报告共22篇,报道病例122例。X-CGD患儿首次发病年龄中位数为1月,确诊年龄中位数为1岁6月;X-CGD早期临床症状不典型,早期诊断困难,55.3%的患儿从发病到确诊需要一年以上时间。X-CGD患儿急性感染期血清标志物表现为三联征:①白细胞总数明显升高以中性粒细胞升高为主,并伴有不同程度的贫血;② CRP显著升高;③ 体液免疫表现为IgG异常增高(>12 g/L)。结论X-CGD患儿发病年龄早,早期临床表现缺乏特异性,导致早期确诊困难;X-CGD患儿在急性感染期血清标志物呈典型的三联征,可为早期确诊提供线索。
X-连锁慢性肉芽肿病;血清学标志物;临床特点;早期诊断;儿童
(ChinJLabDiagn,2017,21:1727)
慢性肉芽肿病(CGD)是一种少见的吞噬细胞功能缺陷所致的原发性免疫缺陷病,美国发病率为1∶200,000-1∶250,000[1,2]。发病机理为吞噬细胞内还原型辅酶Ⅱ(NADPH)复合物功能缺陷,细胞无法正常产生超氧离子和过氧化氢,失去杀伤过氧化物酶阳性细菌与真菌的能力。其中CYBB基因突变所致X-连锁慢性肉芽肿病(X-CGD)最为常见,占所有患者的65%,男性患儿发病,其母亲为携带者。患儿可表现为反复发生的细菌和真菌感染,在感染部位形成肉芽肿。2/3患儿首次发病年龄早,甚至新生儿期起病[3],临床表现缺乏特异性,早期确诊难度大。确诊所需的特殊检查,国内只有少数几家医院开展,基因诊断耗费时间又长。待诊断明确时,感染往往已累及多个系统,甚至危及生命。由于本病为罕见病,临床上大样本报道不多,大部分为单个或数个病例报道,难以从单中心数据中总结和发现特征。为了能在临床上更早的发现、识别此疾病,我们总结了中国X-CGD患儿的临床特征和早期血清检查指标,并进行meta分析,找到了X-CGD患儿早期血清生物标志物特点,以期为早期诊断提供线索。
1 研究方法
1.1文献检索
以 “慢性肉芽肿病”或“X-连锁慢性肉芽肿病”为检索词,在万方数据库、CNKI数据库以及pubmed数据库中检索2017年6月前发表的有关中国儿童X-CGD的相关临床文献,主要为病例报告,重点关注文献中已经标明患儿临床特点和血清相关标志物者,并甄别文献进行去重、汇总和分析。
1.2文献纳入和排除标准
为了总结X-CGD患儿的临床特征和血清生物学特征,文献检索时文献纳入标准如下:① 必须是关于中国儿童明确诊断的X-CGD文献和病例报道;② 文献中有明确的临床表现描述,如发病年龄,初次发病临床表现,确诊年龄及确诊时临床表现;③ 文献中有明确关于血清生物学标志物的记录如血象、CRP、免疫功能等。排除(剔除)标准如下:① 单纯基因突变报道或其他报道而无临床描述和(或)血清生物学标志物记录者;② 同一作者或数篇有同一作者的文献,经仔细核对发现病例被重复使用者,剔除重复病例;③ 其他类型的CGD,而非X-CGD者。
1.3统计分析
对发病年龄、确诊年龄等数据进行正态分布分析,符合正态分布者,采取均值±标准差表示;不符合正态分布者,采取中位数及25%和75%可信区间表示;临床表现按受累器官或系统进行列表分析,并计算百分比进行描述性分析。
2 研究结果
2.1病例资料及发病、确诊年龄比较
检索已发表文献包括病例报告共37篇,经过纳入和排除标准筛选后得到文献22篇,报道病例122例,均为男性患儿,见表1。85例患儿记录了明确的发病年龄和确诊年龄,大部分患儿发病年龄早,确诊晚。患儿首次发病年龄和确诊年龄分布见图1,发病年龄中位数1月(新生儿期-3月),74例患儿生后3个月内发病(占87.1%),其中39例新生儿期起病,占45.9%;仅9例1岁以后起病,占10.6%。确诊年龄中位数为1岁6月(新生儿期-3岁),生后3月内确诊者仅21例(24.7%),这些患儿大多来自北京、上海、重庆等儿科综合实力强的医院,46例1岁后确诊,占54.1%,其中3岁后确诊者19例,占22.4%。发病到确诊时间差中位数为14个月(0-32月),起病后3个月内得到明确诊断的患儿仅26例,占30.6%,大部分患儿(47例,占55.3%)从发病到明确诊断需要一年以上时间,见图2。患儿在确诊之前,往往因反复感染而多次住院治疗。
表1 纳入文献和病例数列表
2.2患儿首次发病及确诊受累系统比较
X-CGD患儿起病早,早期临床症状不典型,见图3。在有明确记录的25例中,以肺炎(12例,占48%)、皮肤脓疱疹(8例,占32%)、发热(6例,占24%)、肛周脓肿(5例,占20%)及脓毒症(4例,占16%),最为多见,上述疾病亦是婴幼儿常见感染性疾病,缺乏特异性,很难为早期诊断提供线索。而在婴幼儿期不常见疾病如淋巴结结核(3例,占12%)、肺结核(2例,占8%)等疾病,虽可为早期诊断提供线索,但在X-CGD患儿首次发病中发病率低,帮助有限。
X-CGD首次发病的症状不典型,受累系统不多,确诊需要经历较长时间,待明确诊断时患儿已经出现多个系统受累,甚至危及生命,见图4。在有记录的80例患儿中,肺炎甚至真菌性肺炎仍是最常见疾病(62例,占77.5%),其次为淋巴结结核(25例,占31.3%)、肺结核(21例,占26.3%)和肛周脓肿(20例,占25%),甚至部分患儿合并危及生命的严重疾病如化脓性脑膜炎(5例,占6.3%)、肝脓肿(7例,占8.75%)等。


2.3患儿急性期就诊血生物标志物特点
患儿发病早期临床表现缺乏特异性,尤其是新生儿期起病患儿,往往没有典型的肉芽肿表现,给诊断带来了很大困难,因此我们总结文献报道病例发现X-CGD患儿在急性感染期存在如下生物标志物特点,可为早期诊断提供线索。见表2。
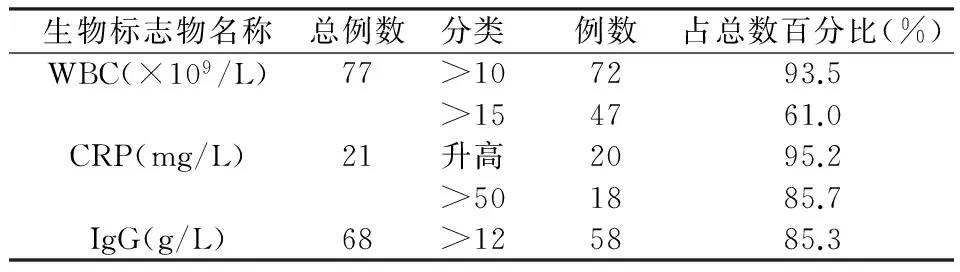
表2 X-CGD患儿血清生物标志物三联征统计
2.3.1白细胞总数增高,以中性粒细胞增高为主,伴不同程度贫血 在确诊的122例患儿中,77例记录了白细胞水平,72例患儿(占93.5%)表现为白细胞总数增高(>10×109/L),以中性粒细胞增高为主(占100%),其中白细胞总数>15×109/L者占47例,占所有患儿61.0%,占白细胞总数增高者中65.3%。大部分患儿除白细胞总数增高外,伴有不同程度贫血。有记录13例中11例有贫血,占84.6%。
2.3.2急性感染期,血CRP异常增高 患儿在急性感染期,除血象增高外,还表现为CRP异常增高,122例患儿中21例记录了CRP水平,其中20例有CRP增高,占有记录者95.2%。其中CRP >50 mg/L以上者18例,占有记录者85.7%。
2.3.3免疫球蛋白IgG水平异常增高 患儿在急性期除血象、CPR显著增高外,还表现为免疫球蛋白IgG水平异常增高,患儿在未输注丙种球蛋白的前提下,血IgG水平异常增高。共68例记录了血IgG水平,其中IgG显著增高者(>12 g/L)58例,占85.3%。
3 讨论
CGD是一种罕见的原发性免疫缺陷病,患者通常在出生后数月出现临床表现,大多数CGD患儿在出生后第一年至少会发生一次严重的感染,其中肺炎最为常见,但早期的临床表现不具备特异性给早期诊断带来困难[4-6]。在我国,原发性免疫缺陷病的筛查尚未纳入国家法定筛查项目及临床医生对该病的早期认识不足是导致患儿诊断延误的两个重要因素。 X-CGD是CGD中最常见的临床类型,我们总结我院确诊的10例患儿的临床特征的同时结合我国学者发表的文献,进一步总结我国X-CGD患儿的临床特点和血清学特征,为早期确诊提供帮助。
X-CGD患儿发病年龄早,由于临床医生对该病的早期认识明显不足,导致延误诊断。在有文献报道的85例患儿中,发病年龄中位数为1月,即大部分患儿在新生儿期起病,生后3个月内患病者占87.1%,而确诊时患儿年龄中位数为18个月,在发病后3个月内确诊的患儿仅占24.7%,且早期确诊的患儿大部分来自北京、上海、重庆等综合实力较强的地区和医院。这与医生对该疾病的早期识别能力直接相关。早期快速确诊X-CGD主要依靠呼吸爆发试验,目前能开展此项检查的单位主要集中在这几个地区,也是这些患儿能早期确诊的另一个重要因素。基因诊断是确诊X-CGD的重要诊断手段,但需要耗费一定的时间。目前数据提示患儿自发病到确诊的时间中位数为14个月,即大部分患儿从发病到确诊需要至少一年以上时间,在此期间患儿因反复感染需要多次住院治疗,甚至为了明确诊断,各地奔波就医。基因诊断虽能确诊该类疾病并进行分型,但在我国人群中尚未发现热点突变。 报道显示在过去5年内,仅78.9%的CGD患儿得到明确诊断[7]。因此,急需提高临床医生对该病的早期认识。
CGD属原发性免疫缺陷病,早期接种疫苗可导致患儿疫苗相关性疾病[8]。欧美等国家已经将原发性免疫缺陷病纳入新生儿筛查,我国目前暂未纳入法定新生儿筛查。X-CGD患儿临床表现呈现一定的特征性。患儿首次发病年龄早,主要表现为肺炎、皮肤脓疱疮、发热、脓毒症等,这些疾病在婴幼儿中属于常见病和多发疾病,单靠临床症状很难在早期考虑此诊断。而一些可为诊断提供线索的婴幼儿期非常见疾病如肺结核、淋巴结结核、肝脓肿在首次发病中所占比例非常低,未能引起临床医生的足够重视。因此,待患儿明确诊断时,往往已经出现反复的感染并累及多个系统。肺炎仍为这类患儿主要受累疾病,占77.5%,与Wu Jing等报道80%的患儿存在肺部感染相当[9],也与欧美国家相似,如Winkelstein JA等报道肺部感染比例为79%,Vandenberg JM报道为66%[10,11]。其次为淋巴结结核、肺结核,这与欧美国家报道显著不同(均为0%)。主要原因在于我国尚未开展免疫缺陷病相关的新生儿疾病筛查,而卡介苗接种已纳入国家计划免疫接种,因此,这部分存在免疫缺陷的患儿因接种卡介苗而患卡介苗相关性疾病。患儿在出现反复重症感染、甚至淋巴结结核和肺结核这些在免疫功能正常的婴幼儿非常罕见的疾病时,才引起临床医生的重视。而此时患儿往往出现多系统受累,治疗非常困难。因此,有必要在患病早期进行诊断。
我们发现该类患儿虽早期临床表现缺乏特异性,但在感染急性期血清生物标志物呈现典型的三联征,在此基础上结合病史可为早期疑诊提供线索。大多数患者在感染急性期表现为白细胞总数增高,以中性粒细胞增高为主。我们总结患儿中白细胞总数增高(>10×109/L)占93.5%,均以中性粒细胞增高为主(100%),其中白细胞总数>15×109/L者占所有有记录患儿61.0%,占白细胞总数增高者中65.3%。细菌或真菌感染后,正常吞噬细胞会吞噬细菌或真菌,并启动“呼吸爆发”,最终清除感染原,而X-CGD患儿因基因缺陷导致NADPH氧化酶复合物功能异常,“呼吸暴发”受损,吞噬细胞不能有效的杀灭微生物,反馈性引起骨髓产生更多的吞噬细胞,释放入血。故患儿出现持续性血象增高,以中性粒细胞增高为主。长期持续白细胞增高及感染清除障碍,影响患儿营养摄入,患儿除白细胞总数增高外,伴不同程度贫血。 在有记录的13例中11例有不同程度的贫血,占84.6%。X-CGD患儿除血象增高外,第二个血清学特征为CRP呈现较高水平的增长。21例记录了CRP水平,其中20例有CRP增高,占有记录者95.2%。CRP水平>50 mg/L以上者18例,占有记录者85.7%。由此可以看出,炎症指标的显著增高,是CGD患儿在感染急性期的显著特征。第三个特征为血IgG水平异常增高。在68例记录了血IgG水平病例中,IgG显著增高者(>12 g/L)58例,占85.3%。在既往的研究中也发现CGD患儿有90%的高丙种球蛋白血症[12]。患儿在无丙种球蛋白输注病史的基础上,出现IgG的显著增高,提示患儿存在免疫功能异常,也是X-CGD早期疑诊的线索之一。也有文献报道部分CGD患儿存在血清IgE增高[13]。此三项血清学指标既属常规生化指标,亦为感染患儿常规检查指标,目前全国大部分医院均已开展,便于临床医生甚至基层医生早期识别。
干细胞移植是根治X-CGD的唯一有效方法[14,15],CRISPR-Cas9基因治疗尚处在探索阶段[16]。有研究表明rhIFN-γ和复方磺胺甲噁唑、伊曲康唑连用,可增强患儿的免疫能力,减少重症感染的发生率,改善X-CGD患儿的预后[17],而另一项长期随访发现rhIFN-γ不仅不能降低感染的发生率甚至可能出现相关副作用如发热、头痛等[18]。因此,早期诊断,避免感染是预防本病继续进展的关键。
综上所述,X-CGD患儿在早期临床表现缺乏特异性,但在感染急性期,存在炎症指标的显著升高,伴随不同程度的贫血,免疫球蛋白IgG水平异常增高,有此三联征存在再结合患儿临床表现,可为早期诊断提供线索。
[1]Holland SM.Chronic granulomatous disease[J].Hematol Oncol Clin North Am,2013,27(1):89.
[2]Mahdaviani SA,Mohajerani SA,Rezaei N,et al.Pulmonary manifestations of chronic granulomatous disease[J].Expert Rev Clin Immunol,2013,9(2):153.
[3]Segal BH,Romani L,Puccetti P.Chronic granulomatous disease[J].Cell Mol Life Sci,2009,66(4):553.
[4]Gardiner GJ,Deffit SN,McLetchie S,et al.A role for NADPH oxidase in antigen presentation[J].Front Immunol,2013,4:295.
[5]Roos D,de Boer M.Molecular diagnosis of chronic granulomatous disease[J].Clin Exp Immunol,2014,175(2):139.
[6]Rawat A,Vignesh P,Sharma A,et al.Infection Profile in Chronic Granulomatous Disease: a 23-Year Experience from a Tertiary Care Center in North India[J].J Clin Immunol,2017,37(3):319.
[7]Huang Xu,Wen Tian Lu-ying Zhang,et al.Clinical and Molecular Features of 38 Children with Chronic Granulomatous Disease in Mainland China[J].J Clin Immunol,2014,34(6):633.
[8]Ying W,Sun J,Liu D,et al.Clinical characteristics and immunogenetics of BCGosis/BCGitis in Chinese children: a 6 year follow-up study[J].PLoS One,2014,9(4):e94485.
[9]Wu J,Wang WF,Zhang YD,et al.Clinical Features and Genetic Analysis of 48 Patients with Chronic Granulomatous Disease in a Single Center Study from Shanghai,China(2005-2015):New Studies and a Literature Review[J].J Immunol Res,2017,2017:8745254.
[10]Winkelstein JA,Marino MC,Johnston RB Jr,et al.Chronic granulomatous disease.Report on a national registry of 368 patients[J].Medicine (Baltimore),2000,79(3):155.
[11]van den Berg JM,van Koppen E,Ahlin A,et al.Chronic granulomatous disease: the European experience[J].PLoS One,2009,4(4):e5234.
[12]Wolach B,Gavrieli R,de Boer M,et al.Chronic granulomatous disease: Clinical,functional,molecular,and genetic studies.The Israeli experience with 84 patients[J].Am J Hematol,2017,92(1):28.
[13]Patiroglu T,Gungor HE,Lazaroski S,et al.Chronic granulomatous disease with markedly elevated IgE levels mimicking hyperimmunoglobulin E syndrome[J].Acta Microbiol Immunol Hung,2013,60(2):155.
[14]Norman M,David C,Wainstein B,et al.Haematopoietic stem cell transplantation for primary immunodeficiency syndromes: A 5-year single-centre experience[J].J Paediatr Child Health,2017,doi: 10.1111/jpc.13643.
[15]唐湘凤,卢 伟,井远方,等.非血缘脐血干细胞移植治疗X连锁慢性肉芽肿7例[J].中国小儿血液与肿瘤杂志,2016,21(5):231.
[16]De Ravin SS,Li L,Wu X,et al.CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease[J].Sci Transl Med,2017,11,9(372).
[17]Goldblatt D.Recent advances in chronic granulomatous disease[J].J Infect,2014,69(Suppl 1):S32.
[18]Martire B1,Rondelli R,Soresina A,et al.IPINET.Clinical features,long-term follow-up and outcome of a large cohort of patients with Chronic Granulomatous Disease:an Italian multicenter study[J].Clin Immunol,2008,126(2):155.
SerumBiomarkerscanbeusedforEarlydiagnosisinChildrenwithX-LingkedChronicGranulomatousDiseaseandaLiteratureReview
CHENShao-hua,YINGYan-qin.
(DepartmentofPediatrics,TongjiHospital,TongjiMedicalCollege,HuazhongUniversityofScienceandTechnology,Wuhan430030,China)
ObjectiveTo explore the clinical features and early biomarkers of X-linked chronic granulomatosis disease(X-CGD) in children and to provide clues for early diagnosis.MethodsA total of 122 cases of X-CGD children were enrolled in this study from 22 papers by literature searching,and we summarized the clinical features and serum biomarkers by meta analysis.ResultsThe median onset age was 1 month,and the median age at diagnosis was 1 year and a half.Since X-CGD had no specific clinical symptoms at early stage,early diagnosis was difficult.55.3% patients got their diagnosis lasted more than one year after the onset of first symptoms.X-CGD children at acute infection showed three signs of serum biomarkers:① The total number of white blood cells increased significantly,especially the N%, accompanying with some degrees of anemia; ② CRP increased significantly; ③ Immune systems showed high IgG levels(> 12 g/L).ConclusionIt is difficult to diagnosis X-CGD at early age because of nonspecific clinical features.we found X-CGD children suffering acute infection showed typical three signs of serum biomarkers,which can provide clues for early diagnosis.
X-linked chronic granulomatosis disease(X-CGD); serum biomarkers; clinical features; early diagnosis; children
R593.31
A
2017-07-29)
*通讯作者
1007-4287(2017)10-1727-05
