Intermolecular Interactions of 3,6-Bis-nitroguanyl- S-tetrazine Dimers: A Density Functional Theoretical Calculation
2017-11-01HUYinNINGYanliKANGYingSONGJirongMAHaixia
HU Yin,NING Yan-li,KANG Ying,SONG Ji-rong,MA Hai-xia
(1. Xi′an Modern Chemistry Research Institute, Xi′an 710065, China;2. School of Chemical Engineering,Northwest University, Xi′an 710069, China;3. Conservation Technology Department, the Palace Museum, Beijing 100009, China)
IntermolecularInteractionsof3,6-Bis-nitroguanyl-S-tetrazineDimers:ADensityFunctionalTheoreticalCalculation
HU Yin1,NING Yan-li1,KANG Ying1,SONG Ji-rong2,3,MA Hai-xia2
(1. Xi′an Modern Chemistry Research Institute, Xi′an 710065, China;2. School of Chemical Engineering,Northwest University, Xi′an 710069, China;3. Conservation Technology Department, the Palace Museum, Beijing 100009, China)
Nine fully optimized geometries and electronic structures on potential energy surface of 3,6-bis-nitroguanyl-S-tetrazine (DNGTz) dimers have been obtained with density functional theoretical (DFT) method at the B3LYP/6-31G*level. The intermolecular interaction energy was calculated with zero point energy (ZPE) correction and basis set superposition error (BSSE) correction. The greatest corrected intermolecular interaction energy of the dimers is -62.24kJ/mol. Natural bond orbital (NBO) analysis is performed to reveal the origin of the interaction. Based on the vibrational analysis, the changes of thermodynamic properties from the monomer to dimer with the temperature ranging from 200.0K to 800.0K have been obtained using the statistical thermodynamic method. It was found that the dimerization are mainly contributed by the strong hydrogen bonds, while the binding energies are not only determined by hydrogen bonding. The dimerization process of dimers I, III, IV, V and VII can spontaneously occur at 200K, which indicates that these dimers can be stably present at room temperature.
high-nitrogen energetic material; 3,6-bis-nitroguanyl-S-tetrazine (DNGTz); intermolecular interaction; density functional theory (DFT); natural bond orbital (NBO) analysis; thermodynamic property
Introduction
High-nitrogen energetic materials have superior chemical properties and detonation performance compared with the traditional energetic materials (EMs)[1]. Their molecular structures contain a large amount of N—N and C—N bonds, with high enthalpy of formation, low sensitivity and high thermal stability[2]. Moreover, high nitrogen content makes them present high density and easily obtain oxygen balance, thus, the main combustion product is clean gas (N2)[3]. Tetrazine compounds are the typical representative of the high nitrogen energetic compounds. The formula of tetrazine ring is C2H2N4, therefore the nitrogen content can reach 68.3%, which make tetrazine compound be an ideal group for the development of new EMs[4].
The application of tetrazine compounds can almost involve in solid propellants, new type high-energy insensitive explosives, and civil combustible-gas generators[5-10]. In recent years, there are more reports about synthesis and characterization of tetrazine compounds[11-12].
3,6-Bis-nitroguanyl-S-tetrazine is an important high nitrogen heterocyclic compound[13]with nitrogen content, enthalpy of formation and density of 58.7%, +389kJ/mol[14]and 1.76g/cm3, respectively. However, the enthalpy of formation of nitroguanyl is -98.7kJ/mol, which indicates that the introduction of tetrazine ring can greatly increase the enthalpy of formation of EMs. According to DSC analysis, DNGTz decomposes at 228℃ with the peak temperature of 269 ℃, higher than that of a traditional explosive of RDX (1,3,5-Trinitrohexahydro-1,3,5-triazine)[15]. Its impact sensitivityH50is 65cm, friction sensitivity is higher than 36kg (BAM) and static inductance degree is higher than 0.36J[16], which indicates that DNGTz is more stable than RDX[17-21]. At present, scientists have studied the synthesis, combustion performance of DNGTz[22]and found that DNGTz is a prospective explosive, however, its theoretical calculation is little done.
It is known that intermolecular interaction could predict the binding force of the molecule and the stability of a system. And many physical, chemical and detonation properties of explosive are related to their state of aggregation. Therefore, there is an important academic and application significance to carry out studies on the intermolecular interactions of EMs[23-31]. In this study, we theoretically investigated the intermolecular interaction for the DNGTz dimers. Its nine fully optimized geometries, electronic structure, the binding energy and thermodynamic properties in the dimerization process were analyzed. Natural bond orbital (NBO) on the DNGTz dimers were also analyzed to explore the source of the interactions, which provides basic data for the structure-property relationship of this compound for further research.
1 Computational Methods
The initial structures of DNGTz monomers and dimers obtained from CHEM3D software were fully optimized at the DFT-B3LYP level by the Berny method[32]with 6-31G*basis set to obtain the geometries of the monomer and all possible optimized dimers. And in order to determine the suitability of the basis set, single-point calculation were performed on the geometries optimized by the B3LYP/6-31G*with B3LYP/6-311++G**basis set. The intermolecular interaction energies of the studied dimers were corrected both with basis set superposition error (BSSE) and zero point energies (ZPE). The interaction energies were compared with the above methods. Finally, NBO analysis and frequency calculations were carried out on each optimized structure. Thermodynamic data and their changes upon dimerizing were derived from statistical thermodynamics based on the frequency calculation. All these calculations have been carried out with the Gaussian 98[33]program using the default Gaussian convergence criteria.
2 Results and Discussion
2.1Optimizedstructure
The stable structures of DNGTz monomer and the nine dimers were obtained (Fig.1). The geometrical parameters of the monomer and the dimers are collected in Table 1.
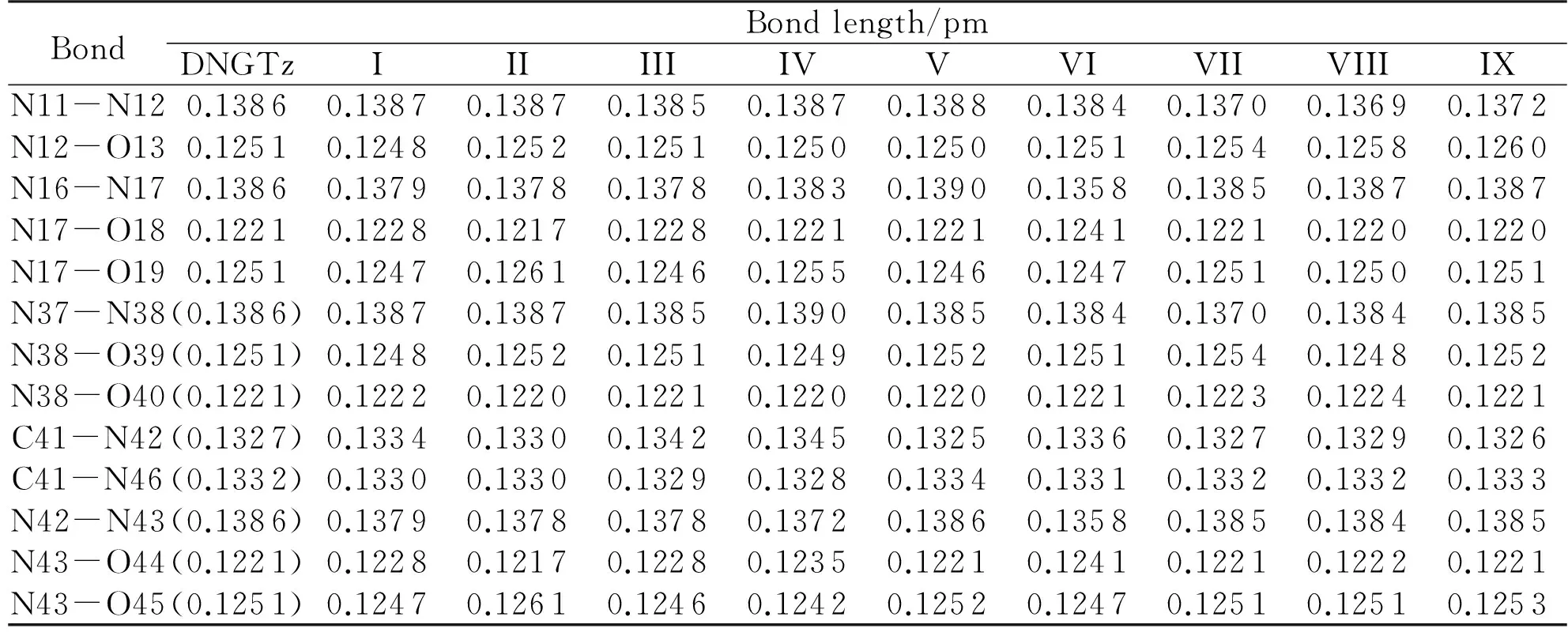
Table 1 The Bond lengths of DNGTz and (DNGTz)2 at B3LYP/6-31G* level
Note: values in parentheses are data of the monomer
Compared to the monomer, the changes of bond lengths for the nine dimers mainly occur in the near of hydrogen bond (Table1). Apart from dimers III and V, the changes of bond lengths for other dimers are similar: if there is an apparent increase of one bond, there must be an apparent decrease of another bond. The bond lengths of N16—N17 and N42—N43 lengths of dimer I decrease by 0.7pm, while those of N17—O18 and N43—O44 increase by 0.7pm. The N16—N17 and N42—N43 lengths of dimer II decrease by 0.8 pm, while the N17—O19 and N43—O45 increase by 1.0pm. The N42—N43 bond length of dimer IV reduces by 1.4pm, and then the N43-O44 length rises by 1.4pm. The bond lengths of N16—N17 and N42—N43 of dimer VI decrease by 2.8pm, while those of N17—O18 and N43—O44 increase by 2.0pm. The lengths of N11—N12 and N37—N38 of dimer VII decrease by 1.5pm, while the lengths of N12—O13 and N38—O39 increase by 0.3pm. The N11—N12 length of dimer VIII decreases by 1.7pm, while the N12—O13 length increases by 0.7pm. The N11—N12 length of dimer IX decreases by 1.4pm, while the N12—O13 length increases by 1.0pm. The X-NO2(X = N, C or O) bond in nitro explosives is generally regarded as the detonating trigger bond[34-35]. In dimers I, II, IV, VI, VII, VIII and IX, there are at least one interaction between the amino group of one monomer and the X-NO2group of another monomer, which shortens the N-N bond near the X-NO2group, this phenomenon is most apparent in dimer VI. Therefore it can be inferred that the intermolecular interaction can reduce the sensitivity of DNGTz. The changes of bond angle are within 5.94° of the nine structures of DNGTz dimers compared to those of monomer molecules. The dihedral angles of dimers I, VI and VIII almost have no change, therefore the atoms still maintain the planarity of the molecule. While some dihedral angles of dimers II, III, IV, V, VII and IX change greatly. TheD(C9-N11-N12-O13),D(C9-N11-N12-O14),D(C35-N37-N38-O39) andD(C35- N37-N38-O40) of dimer VII decrease by 16.56°, 14.49°, 16.54° and 14.47°, respectively, which implies that the rotation has occurred in the -NO2group adjacent to the hydrogen bond in dimer VII. Similar analysis shows that obvious rotation has occurred in the nitro or guanidino groups adjacent to the hydrogen bond in dimers III, IV and V. In dimers II and IX, the rotation occurs in the -NO2groups adjacent to the hydrogen bond.
Fig.1 shows that there are six, four and three hydrogen bonds in dimers I, II and IV, respectively, while there are two H-bonds in other dimers. Generally speaking, the binding energies (the negative value of the intermolecular interaction energy) usually determine by the H-bonding lengths when the intermolecular contacts are similar. Here it is determined by the strength of the intermolecular hydrogen bond. It can be speculated that the intensities of binding energy and stability may be in the order: III>VI>V>VII>IX>VIII.
2.2Interactionenergy
Both the uncorrected and corrected binding energies obtained for all the dimers are listed in Table 2. Generally speaking, the BSSE will be higher with a smaller basis set. The BSSE is larger for dimers at the B3LYP/6-31G*level (Table 2), so BSSE values were corrected by BSSE×50%[36].

Table 2 Zero point energy and binding energies(kJ/mol) at the B3LYP/6-31G* level
Table 2 shows that the uncorrected interaction energy |ΔE| may be in the order: III>I>VI> IV>V>VII>II>VIII>IX and the correction interaction energy (|(ΔE)C,ZPEC|) after BSSE and ZPE may be in the order: III>I>VI>IV>V>VII >IX>VIII>II. For the order of IX, VIII an II, it is not so consistent because the binding energies of these three dimers are close to each other, these values can be easily affected by the basis set we selected. The largest correction interaction energy after BSSE and ZPE for DNGTz dimers is -62.24kJ/mol which belongs to the dimer III. The binding energy for each hydrogen bond is -31.12kJ/mol,which is much higher than the best experimental estimate of the water dimer dissociation energy (15kJ/mol)[37], indicating that the bingding energy for each hydrogen bonding is strong. This is in consistence with the intensities of stability which is determined by the bonding lengths and the intermolecular contacts distance for the dimers. Thus, the H-bonding plays an important role in the intermolecular interactions.
In order to determine the suitability of the basis set, single-point calculation were performed on the geometries optimized by the B3LYP/6-31G*with B3LYP/6-311++G**. The results show that the BSSE corrected binding energies with different basis sets give a different stability order and the correction interaction energy (|(ΔE)C|) after BSSE may be in the order: III>I>VI>IV> V>VII>VIII>IX>II.
2.3Atomicchargesandchargetransfer
Table 3 lists the natural bond orbital (NBO) atomic charges of the monomer and the dimers. Compared to the monomer, the changes of charge in all the dimers (Table 3 and Fig.1) mainly occur on the O…H and N…H atoms. In dimer VII, the charges on the N10, N36, H24 and H50 increase by 0.0063-0.0118e, while the charges on O13 and O39 decrease by 0.0231e. In dimer VIII, the charges on the N11 and N28 increase by 0.0110 and 0.0161e, while the charges on O13, O14, N29 and O40 reduce by 0.0074-0.0209e. In dimer I, the charges on the N2, N28, C4, C30, N10, N36, O19 and O45 increase by 0.0125-0.0246e, while the charges on N3, N29, N7, N33, O18 and O44 decrease by 0.0108-0.0510e. In dimer VI, the charges on the N3, N29, N16, N42, O19 and O45 increase by 0.0165-0.0257e, while the charges on N7, N33, O18 and O44 reduce by 0.0600-0.0100e. Therefore, due to the intermolecular interaction, if the charge of one oxygen atom in NO2increases, the charge on another oxygen atom will decrease. There is no net charge transfer between N33-H47 and N7-H21 because these two bonds have an opposite charge transfer direction. Mostly, the net charge transfer between the two sub-systems is slight. The dipole moments of DNGTz monomer and nine dimers are 0, 0.0023, 2.8945, 0.3628, 10.7599, 1.6143, 0.1063, 0.2117, 2.7812 and 1.4733 Debye, respectively.
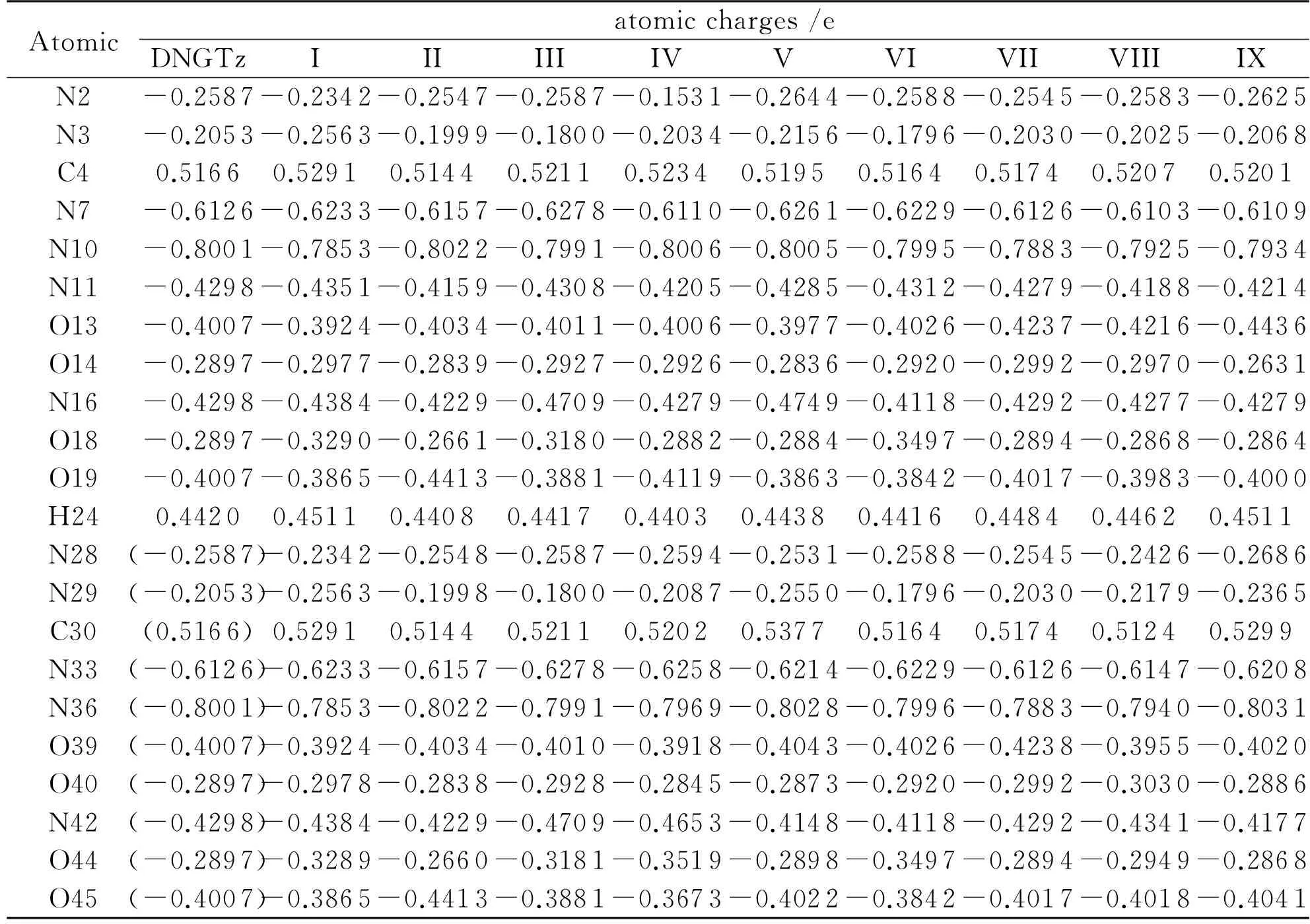
Table 3 The calculated natural atomic charges (e) of DNGTz and (DNGTz)2 at the B3LYP/6-31G* level
Note: Values in parentheses are data of the monomer.
2.4Naturalbondorbitalanalysis
In order to investigate the nature of the intermolecular interactions of (DNGTz)2NBO, We performed the analysis on the DNGTz monomer and its dimers by using the B3LYP/6-31G*. Table 4 summarizes the electron donor track (i), electron acceptor track (j) and the stabilization energy (E) of them.
This is done by examining all possible interactions between filled (donor) Lewis-type NBOs and empty (acceptor) non-Lewis NBOs, and the second order perturbation theory is used to estimate their stabilization energy[38-40]. The stabilization energiesE(2) are proportional to the NBO interaction. When a donor and an acceptor belong to different submolecules in a cluster, it is called an intermolecular NBO interaction. It is these that explore the origin of the intermolecular interactions.
What as what can be seen from the intermolecular NBO interaction in Table 4, in dimer III, which is the dimer with the strongest hydrogen bond, the lone pair (1) of N16, acts as a donor, interacts with N33-H47 σ antibond, as an accepter, the total stabilization energy is 108.41kJ/mol. The lone pair (1) of N42 interacts with N7-H21 σ antibond, the total stabilization energy of these two NBO interactions are 108.11kJ/mol. The lone pairs (1) of N3 and N29 interact with σ of N33-H47 and N7-H21 antibonds in dimer I, which is the dimer with the weakest hydrogen bond, the sum stabilization energies of these two NBO interactions are 61.92kJ/mol. In dimer VI, the main interaction is between the lone pairs on oxygen atom of -NO2in one submolecule and the N—H antibonds of another submolecule. Similar analysis can be done on other dimers. Therefore, the hydrogen bond is the main intermolecular interaction between the two submolecules.
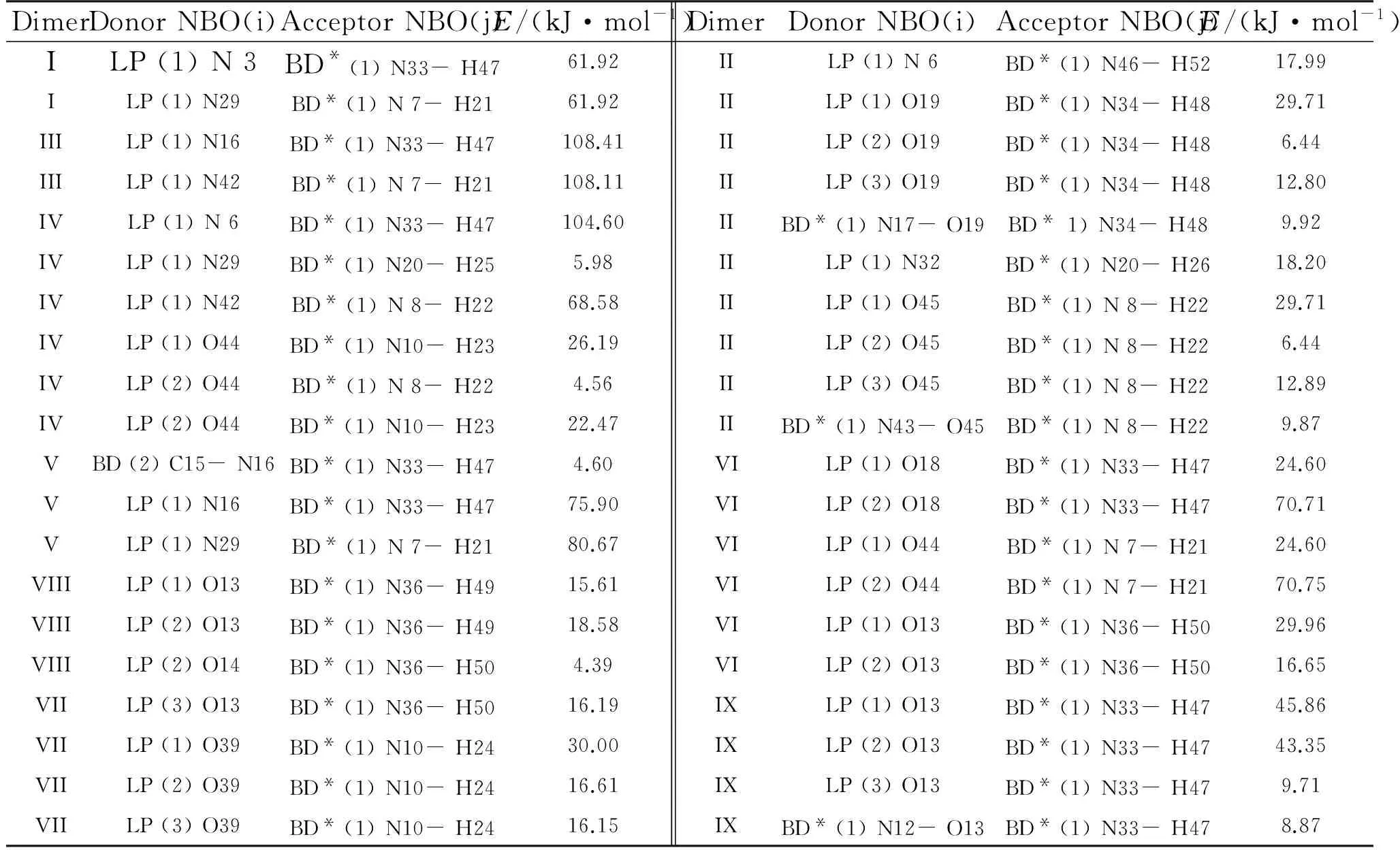
Table 4 Parts of calculated results of (DNGTz)2 at the B3LYP/6-31G* level by NBO analysis
Note: BD denotes bonding orbital; BD*denotes antibonding orbital, LP denotes lone-pair. Only the stable energies over 4.18kJ/mol are listed.
2.5Thermodynamicproperties
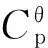
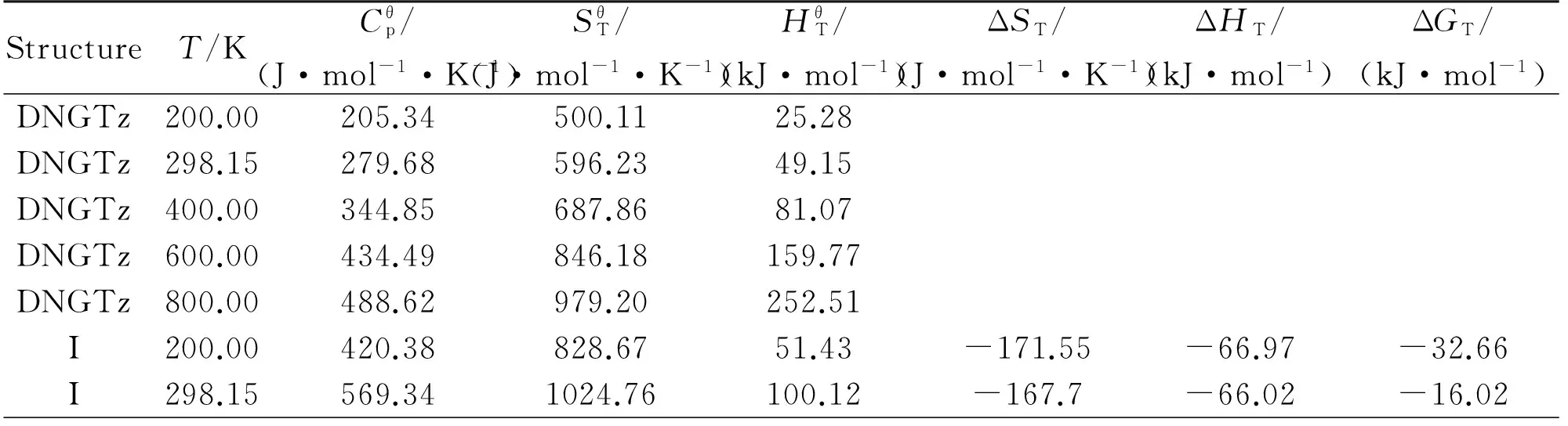
Table 5 The thermodynamic properties of DNGTz and (DNGTz)2 at different temperatures
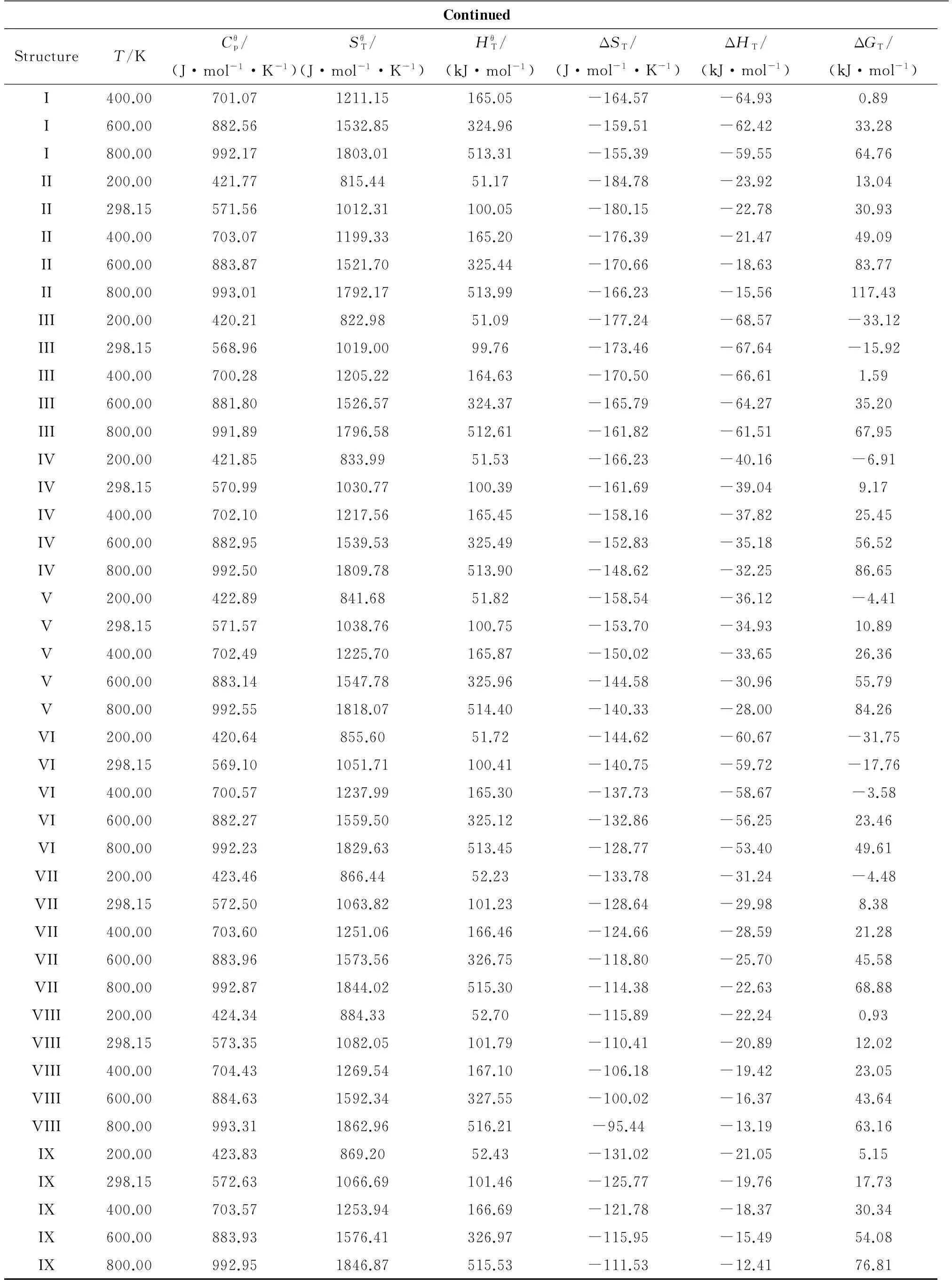
ContinuedStructureT/KCθp/(J·mol-1·K-1)SθT/(J·mol-1·K-1)HθT/(kJ·mol-1)ΔST/(J·mol-1·K-1)ΔHT/(kJ·mol-1)ΔGT/(kJ·mol-1)I400.00701.071211.15165.05-164.57-64.930.89I600.00882.561532.85324.96-159.51-62.4233.28I800.00992.171803.01513.31-155.39-59.5564.76II200.00421.77815.4451.17-184.78-23.9213.04II298.15571.561012.31100.05-180.15-22.7830.93II400.00703.071199.33165.20-176.39-21.4749.09II600.00883.871521.70325.44-170.66-18.6383.77II800.00993.011792.17513.99-166.23-15.56117.43III200.00420.21822.9851.09-177.24-68.57-33.12III298.15568.961019.0099.76-173.46-67.64-15.92III400.00700.281205.22164.63-170.50-66.611.59III600.00881.801526.57324.37-165.79-64.2735.20III800.00991.891796.58512.61-161.82-61.5167.95IV200.00421.85833.9951.53-166.23-40.16-6.91IV298.15570.991030.77100.39-161.69-39.049.17IV400.00702.101217.56165.45-158.16-37.8225.45IV600.00882.951539.53325.49-152.83-35.1856.52IV800.00992.501809.78513.90-148.62-32.2586.65V200.00422.89841.6851.82-158.54-36.12-4.41V298.15571.571038.76100.75-153.70-34.9310.89V400.00702.491225.70165.87-150.02-33.6526.36V600.00883.141547.78325.96-144.58-30.9655.79V800.00992.551818.07514.40-140.33-28.0084.26VI200.00420.64855.6051.72-144.62-60.67-31.75VI298.15569.101051.71100.41-140.75-59.72-17.76VI400.00700.571237.99165.30-137.73-58.67-3.58VI600.00882.271559.50325.12-132.86-56.2523.46VI800.00992.231829.63513.45-128.77-53.4049.61VII200.00423.46866.4452.23-133.78-31.24-4.48VII298.15572.501063.82101.23-128.64-29.988.38VII400.00703.601251.06166.46-124.66-28.5921.28VII600.00883.961573.56326.75-118.80-25.7045.58VII800.00992.871844.02515.30-114.38-22.6368.88VIII200.00424.34884.3352.70-115.89-22.240.93VIII298.15573.351082.05101.79-110.41-20.8912.02VIII400.00704.431269.54167.10-106.18-19.4223.05VIII600.00884.631592.34327.55-100.02-16.3743.64VIII800.00993.311862.96516.21-95.44-13.1963.16IX200.00423.83869.2052.43-131.02-21.055.15IX298.15572.631066.69101.46-125.77-19.7617.73IX400.00703.571253.94166.69-121.78-18.3730.34IX600.00883.931576.41326.97-115.95-15.4954.08IX800.00992.951846.87515.53-111.53-12.4176.81
3 Conclusion
(1) From the DFT calculation, nine fully optimized geometries of DNGTz dimers were obtained. Maximum corrected intermolecular interaction of the most stable dimer III is predicted to be -62.24kJ/mol.
(2) From the geometric analysis, one can find that rotation in the nitroguanidino groups occurred due to the intermolecular interactions. Moreover, the interaction, between the amino group of one monomer and the X-NO2group of another monomer, shortens the N—N bond near the X-NO2group, indicating that the intermolecular interaction can reduce the sensitivity of DNGTz.
(3) The NBO analysis suggests that the net charge transfer in the sub-system between the dimers is small, almost no charge transfer occurs. At last, the dimerization process of dimers I, III, IV, V, VI and VII can spontaneously occur at 200K.
[1] Hu Yin, Ma Hai-xia, Zhang Jiao-qiang, et al. Theoretical study on intermolecular interactions of 3,6- diamino-1,2,4,5-tetrazine dimers[J]. Chemistry, 2010, 73(3): 263-268.
[2] Bai Lin, Hu Yin, Hu Rong-zu, et al. Interaction between 3,6-diamino-1,2,4,5-tetrazine-1,4-di-N-oxide and hydrogen fluoride by DFT[J]. Chinese Journal of Explosives & Propellants(Huozhayao Xuebao),2010, 33(6): 19-24.
[3] Hu Yin, Ma Hai-xia, Li Jun-feng, et al. Density function theoretical study on intermolecular interactions of 3,6-dihydrazino-1,2,4,5-tetrazine dimers[J]. Bulletin of the Korean Chemical Society, 2010, 31(10): 2897-2902.
[4] Li Jun-feng, Ma Hai-xia, Yan Biao, et al. Thermal decomposition of 3,6-bis-nitroguanyl-S-tetrazine by the TG-FTIR[J]. Chinese Journal of Explosives & Propellants(Huozhayao Xuebao), 2010, 33(6): 1-4.
[5] David E C, Damon W P. New heterocycles from tetrazines and oxadiazoles[J]. Journal of Heterocyclic Chemistry, 2009, 46: 88-90.
[6] Hiskey M A, Chavez D E and Naud D L. Propellant containing 3,6-bis(1H-1,2,3,4-tetrazol-5-yl-amino)-1,2,4,5-tetrazine or salts thereof: US,6458227[P], 2002.
[7] Hervé G. Derivatives of 1,1-diamino-2,2-dinitroethene(DADNE) and specific reactivity understanding [J]. Propellants, Explosives, Pyrotechnics, 2009, 34: 444-451.
[8] Shawali A S and Tawfik N M. Novel facile synthesis of imidazo[1,2-b]-[1,2,4,5]tetrazines with potential antimicrobial activity[J]. Arch Pharm Res, 2009, 32(7): 975-982.
[9] Gong Y H, Miomandre F, Meallet-Renault R , et al. Synthesis and physical chemistry of s-tetrazines: which ones are fluorescent and why?[J]. Europen Journal of Organic Chemistry, 2009, 35: 6121-6128.
[10] Steinhauser G, Klapötke T M. Using the chemistry of fireworks to engage students in learning basic chemical principles: a lesson in eco-friendly pyrotechnics[J]. Journal of Chemical Education, 2010, 87(2): 150-156.
[11] Xu Song-lin, Yang Shi-qing, Wang Yun-peng. Research advances in high-nitrogen energetic materials derived from tetrazine[J]. Chemical Propellants and Polymeric Materials, 2007, 5(1): 14-19.
[12] Wang Bo-zhou, Lai Wei-peng, Lian Peng, et al. Novel synthesis, characterization and quantum chemistry study on 3,3′-azobis(6-amino-1,2,4,5-tetrazine) [J]. Chinese Journal of Organic Chemistry, 2009, 29(8): 1243-1248.
[13] Chavez D E, Hiskey M A, Gilardi R D. Novel high-nitrogen materials based on nitroguanyl substituted tetrazine[J]. Organic Letters, 2004, 6(17):2889-2891.
[14] Chavez D E, Tappan B C, Hiskey M A, et al. Novel high-nitrogen materials based on nitroguanyl-tetrazines: explosive properties, thermal decomposition and combustion studies[J]. Propellants, Explosives, Pyrotechnics, 2005, 30(6):412-417.
[15] Badgujar D M. Talawarb M B, Asthana S N, et al. Advances in science and technology of modern energetic materials: An overview [J]. Journal of Hazardous Materials, 2008, 15: 289-305.
[16] Xue Jin-qiang,Shang Bing-kun, Wang Wei, et al.Research advances in tetrazine-based high-nitrogen molecular and ionic energetic compounds[J].Chemical Propellants and Polymeric Materials, 2011, 9(4): 1-8.
[17] Huang Hui, Wang Ze-shan, Huang Heng-jian, et al. Researches and progresses of novel energetic materials[J]. Chinese Journal of Explosives & Propellants(Huozhayao Xuebao), 2005, 28(4): 9-13.
[18] Feng Chang-gen, Zhang Rui, Chen Lang. The cook-off test and its numerical simulation of RDX[J]. Chinese Journal of Energetic Materials, 2004, 12(4): 193-198.
[19] Li Zhi-min, Zhou Ming-rui, Zhang Tong-lai, et al. Study on the electrostatic hazards of lead styphnate[J]. Acta Armamentarii, 2013, 34(8) : 958-964.
[20] Lu Ming, Zhao Sheng-xiang, Chen Jing. Measurement and analysis of the frictional static electricity characteristics of composite RDX[J]. Chinese Journal of Energetic Materials, 2008, 16(6): 708-711.
[21] Wang Gui-xiang, Xiao He-ming, Ju Xue-hai, et al. Theoretical studies on densities, detonation velocities and pressures and electric spark sensitivities of energetic materials[J]. Acta Chimica Sinica, 2007, 65(6):517-524.
[22] Huo Huan,Wang Bo-zhou,Luo YI-fen,et al. Synthesis, characterization and thermal properties of energetic compound 3,6-dinitroguanidino-1,2,4,5-tetrazine (DNGTz) and its derivatives [J]. Journal of Solid Rocket Technology, 2013, 36(4):500-505.
[23] Ma Hai-xia, XIAO He-ming, SONG Ji-rong, et al. Molecular structure of 4-amino-1,2,4-triazol-5-one and a density-functional theoretical investigation of its dimers and crystal band structure[J]. Journal of Chemical Physics, 2008,344(1-2): 79-89.
[24] FANG Guo-yong, Xu Li-na, Hu Xin-gen, et al. DFT study of the interaction between 3-nitro-1,2,4-triazole-5- one and hydrogen fluoride[J]. Journal of Hazardous Materials, 2008, 160(1): 51-55.
[25] Chermahini A N, Ghaedi A, Teimouri A, et al. Density functional theory study of intermolecular interactions of cyclic tetrazole dimers[J]. Journal of Molecular Structure Theochem, 2008, 867(1-3): 78-84.
[26] Ju Xue-hai, Xiao He-ming and Tan Jin-zhi. Theoretical study on intermolecular interactions and thermodynamic properties of imethylnitroamine clusters[J]. Chinese Journal of Chemistry, 2002, 20(7): 629-637.
[27] Xiao H M, Li J S and Dong H S. A quantum-chemical study of PBX: intermolecular interactions of TATB with CH2F2and with linear fluorine-containing polymers[J]. Journal of Physical Organic Chemistry, 2001, 14: 644-649.
[28] Xiao He-ming, Li Jin-shan, Dong Hai-shan. A study on the intermolecular interactions in energetic systems-the mixtures containing NNO2and NH2groups[J]. Chin Acta Chimica Sinica, 2000,58(3):297-302.
[29] Fan Guo-yong, Xu Li-na, Xiao He-ming, et al. Theoretical study on intermolecular interactions of 3-nitro-1,2,4- triazo-5-one with NH3and H2O[J]. Chin Acta Chimica Sinica, 2005, 63(12):1055-1061.
[30] Xu Li-na, Xiao He-ming, Fang Guo-yong, et al. Theoretical study on intermolecular interactions of 3-nitro- 1,2,4- triazo-5-one dimers[J]. Chin Acta Chimica Sinica, 2005, 63(12): 1062-1068.
[31] Li Jin-shan, Zhao Feng, Jing Fu-qian. A theoretical study of intermolecular interaction of HNO3dimer[J]. Journal of Molecular Structure: Theochem, 2001, 574: 213-220.
[32] Zhao Ning-ning, Zhao Ya-li, Hu Yin, et al. Intermolecular interactions and thermodynamic properties of 3,6-diamino-1,2,4,5-tetrazine1,4-dioxide dimers: a density functional theoretical study[J]. South Africa Journal of Chemistry, 2013,66:167-172.
[33] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 98, Revision A[CP/CD]. Pittsburgh: Gaussian, Inc, 1998.
[34] Delpuech A,Cherville J. Relation entre la structure electronique et la sensibilité au choc des explosifs secondaires nitrés-critère moléculaire de sensibilité. I. cas des nitroaromatiques et des nitramines[J]. Propellants, Explosives, Pyrotechnics, 1978, 3(6):169-175.
[35] Xiao H M, Fan J F, Gu Z M, et al. Theoretical study on pyrolysis and sensitivity of energetic compounds. (3) Nitro derivatives of aminobenzenes [J]. Journal of Chemical Physics, 1998, 226:15-24.
[36] Ju Xue-hai, Xiao Ji-jun, Xiao He-ming. Theoretical study on intermolecular interactions and thermodynamic properties of water-hydrogen peroxide clusters [J]. Journal of Molecular Structure (Theochem), 2003, 626(1-3): 231-238.
[37] Feyereisen M W, Feller D, Dixon D A. Hydrogen bond energy of the water dimer[J]. Journal of Physical Chemistry, 1996, 100:2993- 2997.
[38] Reed A E, Weinstock R B and Weinhold F. Natural population analysis[J]. Journal of Chemical Physics, 1985, 83:735-746.
[39] Reed A E and Weinhold F. Natural localized molecular orbitals[J]. Journal of Chemical Physics, 1985, 83:1736-1740.
[40] Reed A E, Curtiss L A, Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint[J]. Journal of Chemical Reviews, 1988, 88(6):899-926.
DNGTz二聚体分子间相互作用的密度泛函理论计算
胡 银1,宁艳利1,康 莹1,宋纪蓉2,3,马海霞2
(1. 西安近代化学研究所,陕西 西安 710065;2.西北大学化工学院,陕西 西安 710069;3. 故宫博物院文保科技部,北京 100009)
在DFT-B3LYP/6-31G*水平下,求得3,6-二硝基胍基-1,2,4,5-四嗪(DNGTz)二聚体势能面上9种优化几何构型和电子结构。用基组叠加误差(BSSE)和零点能(ZPE)校正,计算了分子间相互作用能,二聚体分子间最大相互作用能为-62.24kJ/mol。由自然键轨道(NBO)分析揭示了分子间相互作用的本质。对优化构型进行振动分析,并基于统计热力学求得温度200.0~800.0K从单体形成二聚体的热力学性质变化。结果表明, 二聚主要由强氢键所贡献,而结合能不仅取决于氢键。二聚体I、III、IV、V和VII的二聚过程在200.0K均能自发进行,表明二聚体 I、 III、IV、 V 和 VII 在室温可以稳定存在。
高氮含能材料;3,6-二硝基胍基-1,2,4,5-四嗪(DNGTz);分子间相互作用;密度泛函理论(DFT); 自然键轨道分析(NBO); 热力学性质
TJ55;O64DocumentCodeAArticleID1007-7812(2017)05-0030-09
10.14077/j.issn.1007-7812.2017.05.006
date:2017-02-28;Reviseddate2017-04-28
Foundation:The National Natural Science Foundation of China(No.21673179)
Biography:HU Yin (1984- ),female,engineer,research field:theoretical studies on intermolecular interaction and molecular structure characterization of energetic materials. E-mail: huyin618@163.com
