简化基因测序技术在植物检材个体认定中应用初探
2017-10-24庄艳
庄 艳
(重庆警察学院, 重庆 401331)
简化基因测序技术在植物检材个体认定中应用初探
庄 艳
(重庆警察学院, 重庆 401331)
目的 在涉及中草药真假辨识及个体识别的案件中很大比例的植物类物证都无法用形态学检验方法来鉴别,而植物DNA测序分析技术为我们提供了一种强有力的手段。方法 通过对18个桂花植物样品进行DNA建库和高通量测序、序列多态性分析,评估其遗传多样性,并用基于单核苷酸多态性分析方法探讨了简化基因测序技术用于个体识别的案例。结果 未知植物物证样品A与桂花1的匹配度最高,未知植物物证样品B与桂花4匹配度最高,未知植物物证样品B与桂花11的匹配度最高,该方法能成功识别3个盲测样品。
简化基因测序技术; 单核苷酸多态性; 桂花; 个体识别
0 引言
很多涉及食品安全的案件中常常出现植物类物证,而植物是犯罪现场中一种很容易被忽视的环境和过程证据,大多这些植物类物证都无法用形态学检验方法来鉴别,需要借助植物DNA技术手段,对植物物证的来源和种类进行鉴定,如果能对案件中涉及的植物类材料进行物种鉴别,进而建立起人与物的环境或者过程关系,那鉴定结果对侦查破案和法庭诉讼产生重要的指导和支撑作用[1-3]。
RAD-seq(Restriction Association site DNA sequencing)技术是在第二代测序技术基础上发展起来的,利用限制性内切酶对基因组进行酶切,结合一定大小的插入片段文库,通过高通量测序和信息分析,快速鉴定成千上万的单核苷酸多态性(Single Nucleotide Polymorphism,SNP)信息,获得的SNP位点信息可以较好的代表整个基因组的序列特征,RAD-seq技术操作简单、可简化复杂基因组,而且可以不受参考基因组限制,目前已广泛应用于分子育种、系统进化等领域[4]。但RAD-seq技术应用于涉及植物个体识别的鉴定案例还未见报道。
本文选择没有可参考基因组的植物物证桂花作为研究对象,利用简化基因组测序技术来降低基因组测序和分析的复杂度,构建桂花的个体识别数据库,试图找到能识别桂花同一个体的方法。
1 材料与方法
桂花样品全部与2015年10月采自西南大学校园里,每棵桂花树随机采集2~4片叶片,置于4 ℃冰箱备用,将选取的叶片样品,提取总基因组DNA进行分析。
2 RAD-seq的主要技术流程
2.1 利用限制性内切酶对基因组DNA样品进行酶切,保证产生的RAD标记能够在基因组上均有分布,同时获得的RAD标记数量能够达到实验所需的饱和度。
2.2 建库主要步骤如下:
(1) 用限制性酶消化基因组DNA,并在酶切片段两端加P1接头;(2) 对连接P1 接头的DNA片段进行pooling,并随机打断,收集长度在350~550 bp之间的片段;(3) 在回收片段两端加P2接头。P2接头为分叉的Y 型接头,可阻止未连接P1接头的片段扩增;(4) 选择连接了P1接头的RAD tag进行扩增。
2.3 上机测序
将 PCR 后产物进行DNA片段回收,并对最终构建完成的文库利用安捷伦2100r文库质量检测仪进行检测。RAD-seq测序平台的测序仪器为Illumina Hiseq 4000。
2.4 数据产出
采用Illumina Hiseq4000测序仪对RAD文库进行序列测定。对所测得的原始数据进行过滤,其过滤标准如下:(1)仅使用含有 Hind Ⅲ酶切识别位点Read1序列;(2)reads序列整体质量Q30>85%;(3)如果一条read,它的低质量(Q≤5(E)) 的碱基数占整条read的50%以上,则去掉该reads;(4)去除前5bp不是酶切序列AATTC的reads(5)整条序列中不确定碱基不多于 3 个;(6)去除含有dupulication的reads;(7)去除PolyAreads。过滤后的序列根据index序列划分到具体个体,便于后续分析。
3 结果与分析
3.1 测序基本信息分析
如表1所示,获得15个已知植物物证桂花样本以及3个未知植物物证桂花样本的原始DNA序列,根据如下标准对原始数据进行过滤:(1)仅使用含有Hind Ⅲ酶切识别位点Read1序列;(2)利用Q30标准对序列质量进行评估;(3)所得的序列的前50 bp不存在不确定碱基;(4)整条序列中不确定碱基不多于3个。
15个已知桂花样本的原始数据经过滤后共产出3.47 Gb clean data,3个未知样本的原始数据经过滤后共产出746 Mb clean data。并且G和C含量相近,A和T含量相近,说明测序质量较好。从整体数据量来看,数据有效率高达97.28%。经过滤后,共获得6,638,587,602 bp的Clean data,平均每个个体数据高达368 810 422 bp,数据量统计如表1所示。
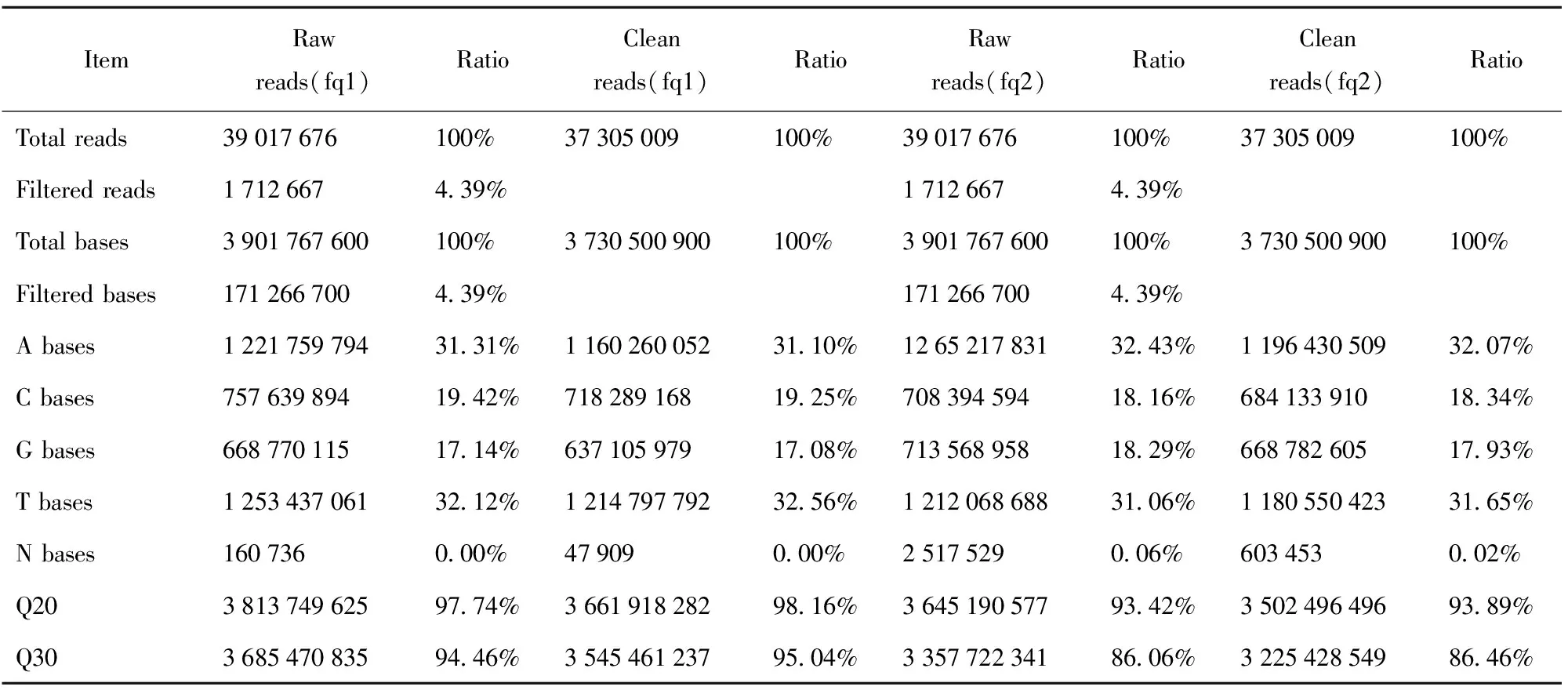
表1 植物物证桂花样本测序质量统计表
3.2 SNP的查找检测和建库
利用RAD技术对过滤后的数据进行聚类和SNP的查找,然后得出初步的SNP的可能位点,且先对15个已知样品通过聚类获得的初始SNP进行初步过滤,得到SNP数量为119 708 5个,过滤标准如下:所有SNP位点总深度须大于等于4,如果SNP为杂合型则次好碱基深度须大于等于2。
如表2所示,植物物证桂花样品获得的SNP数量从3 373到158 467不等,样品平均SNP数量为79 805。Mc Carroll.S.A认为群体遗传上的差异主要是通过对其群体上SNP位点信息,来进行群体间遗传多样性的分析[5]。本实验获得SNP数量最多的是植物物证桂花9,最少的是桂花8。杂合度最高个体是桂花4,高达83.52%。植物物证桂花个体的SNP的差异性反映了个体间的多态性。
3.3 建库比对分析法
为进一步解析系统发育树分析结果的推测,本文进一步通过建立SNP数据库和未知植物物证样品的SNP位点比较来找出具有区分效力的SNP子集,并建立个体识别的分析方法。
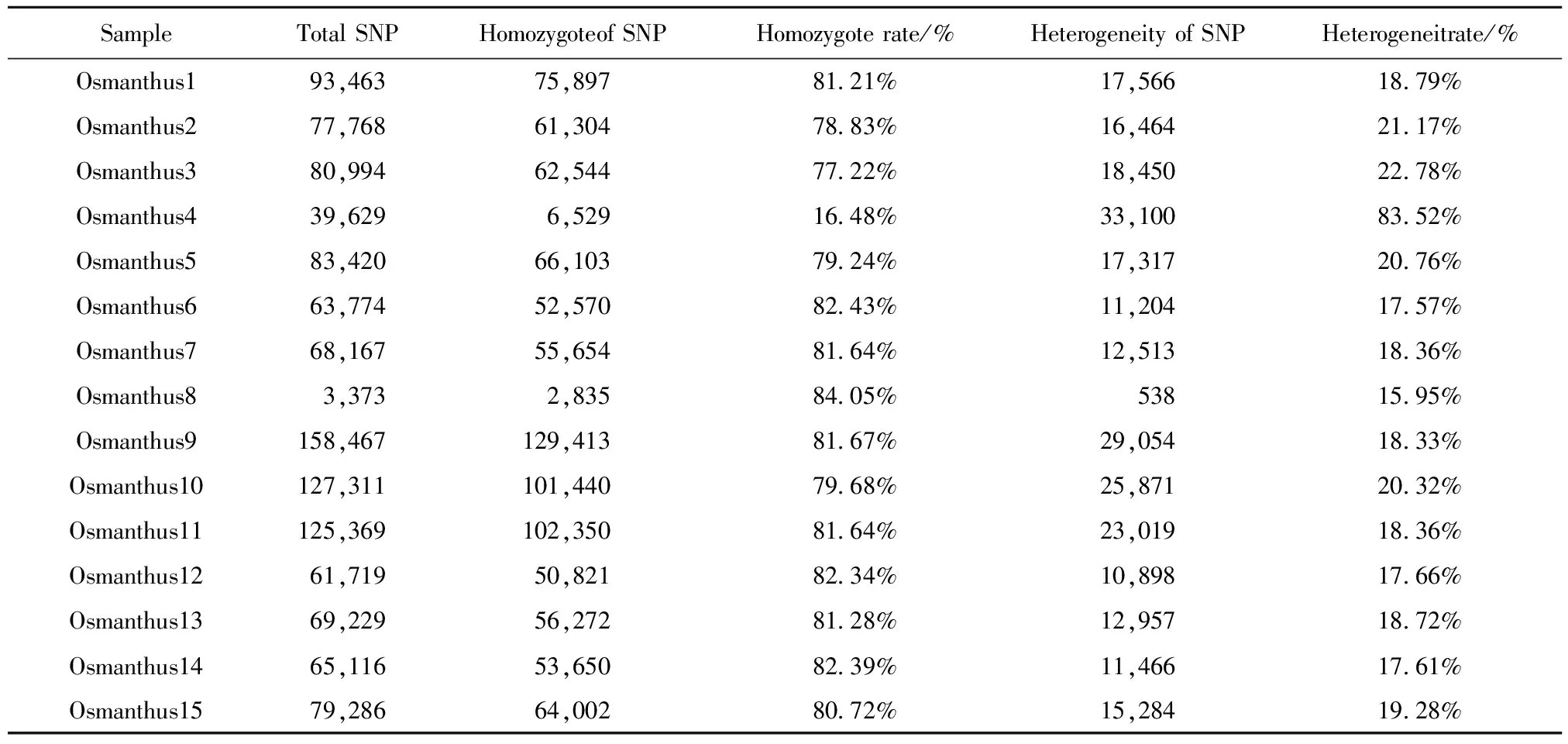
表2 经过滤所得的SNP信息
通过RAD-seq技术获得15个桂花样本的SNP数据集,对此数据集进行筛选(筛选原则:纯合SNP深度至少为4,杂合SNP的次好深度至少为2),将至少在15个样本都存在的136个SNP位点和至少在14个桂花样本都存在1 046个SNP位点,获得 共计1 182个SNP位点,然后用这1 182个SNP数据集建库。
3.3.1 盲测单样品tag簇严格过滤分析
通过RAD-seq技术分别获得3个未知样本的SNP数据集,遵循以上原则进行筛选,然后将得到的SNP位点信息与SNP库做比对,最终得出的统计结果如表3。
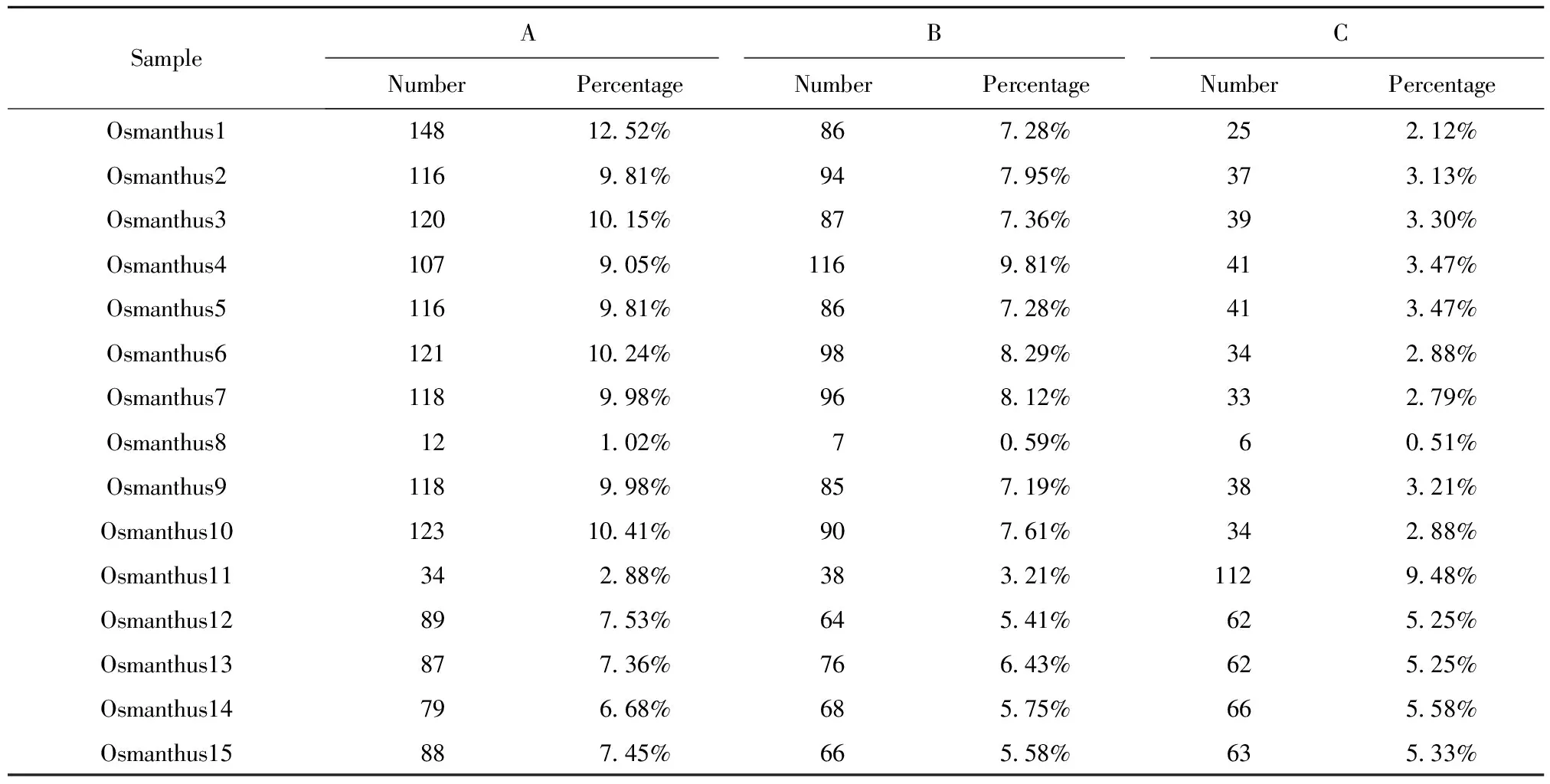
表3 未知样品A、B、C的SNP位点信息与桂花数据库比对的结果
从表3中可以看出,未知植物物证桂花样品A的SNP位点信息与桂花数据库比对后,桂花1与未知植物物证样品A的比对符合度最高,有148个位点符合,符合率达到12.52%;未知植物物证样品B的SNP位点信息与桂花数据库比对后,桂花4与未知样品B的比对的符合度最高,有116个位点符合,符合率达到9.81%;未知植物物证样品C的SNP位点信息与桂花数据库比对后,桂花11与未知样品C的比对的符合度最高,有112个位点符合,符合率达到9.48%。从而推测出A、B、C三个未知植物物证样品分别是桂花1、桂花4、桂花11。分析结果刚好也验证了系统发育树的推测,且与实际情况吻合。
3.3.2 盲测单样品tag簇宽松条件过滤分析
将3个未知植物物证样品的数据产出结果进行聚类,并进行过滤(过滤原则:将tag中不含有N值并且至少有两个tag聚类在一起的tag提取出来),将过滤后的聚类结果与本文15个植物桂花样本所建的SNP库做比对,统计结果如下表4。
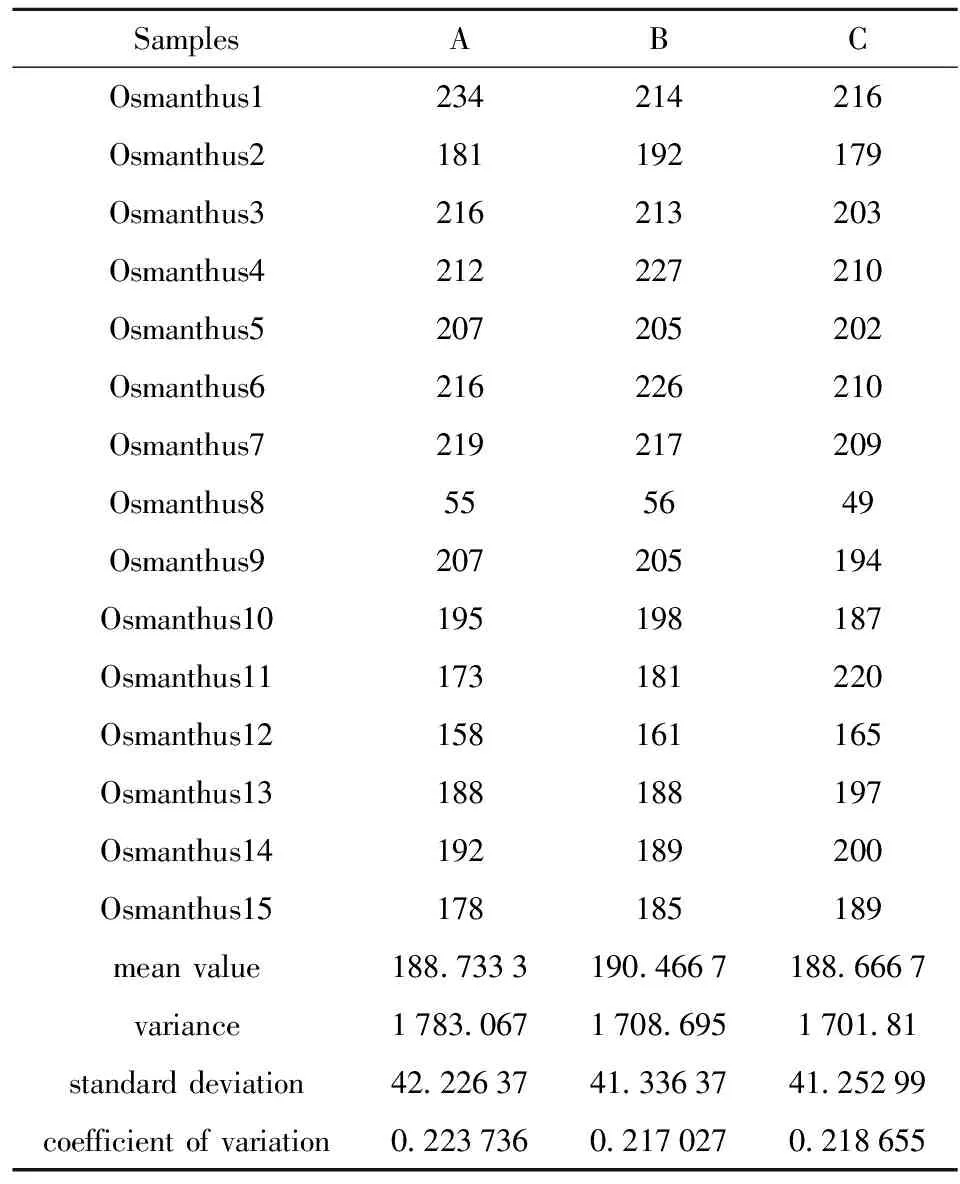
表4 3个未知样品聚类与桂花SNP数据库比对结果
3个未知植物物证样品同时与桂花SNP数据库比对,从中可以近似地看出这样一种情况:未知植物物证样品A与数据库中的桂花1重叠度最高,重叠SNP位点达234,未知植物物证样品B与数据库中的桂花4重叠度最高,重叠SNP位点达227,未知植物物证样品C与数据库中的桂花11重叠度最高,重叠SNP位点达220。这种数据分析结果也很好的验证了系统发育树的推测和盲测单样品tag簇严格过滤分析结果,说明该分析方法是可行且有效的。
4 结果与讨论
在涉及食品安全案件中,如何快速、准确地鉴别中药材的易混品,以及在涉及鉴别植物个体的案件中建立一种行为人及受害者可控物品中附着植物与案件现场的植物是否存在同一性联系的方法就显得尤为重要[6]。本文首次尝试利用RAD-seq技术来识别桂花个体同一性,用未知桂花个体与已知建库中的桂花个体比对分析,得出两个桂花个体的特征点相同,存在同一性联系,这对于查找破案线索,划定侦查范围往往具有重要的意义。
传统的分析方法都是制作系统发育树,通过分析样本之间的亲缘关系来识别3个未知样本。本文的数据采用建库比对分析法来区分未知植物样品,不管是盲测单样品tag簇宽松条件或者是盲测单样品tag簇严格过滤条件的分析结果,可以得到即:未知植物物证样品A与桂花1的匹配度最高,未知植物物证样品B与桂花4匹配度最高,未知植物物证样品C与桂花11的匹配度最高,也就是说,我们鉴定出未知植物物证样品A、B、C对应库中桂花个体1、4和11。这些不同角度的分析方法都进一步说明数据分析的可靠性。
未知植物物证样品A与桂花1有148条SNP位点信息相吻合,符合度达到了12.5%,这12.5%的吻合度是否可以对未知植物物证的样品A与桂花1的同一认定做出定性的判断?在今后的不同物种个体识别的研究中,是否可以建立一个最低的认定标准做定性判断,这些问题还需要大量的样本和实验研究进一步确定。
[1] HEBERT P D N, CYWINSKA A, BALL S L, et al. Biological identifications through DNA barcodes [J].Proceedings of the Royal Society B:Biological Sciences,2003,270(1512):313-321.
[2] 王彦学.植物DNA检验技术在命案现场重建中的应用策略[J].中国司法鉴定,2014(3):53-57.
[3] 王乐,叶健,白雪,等.二代测序技术及其在法医遗传学中的应用[J]. 刑事技术, 2015(5):353-358.
[4] 王洋坤,胡艳,张天真.RAD-seq技术在基因组研究中的现状及展望[J].遗传,2014(1):41-49.
[5] MCCARROLL S A, KURUVILLA F G, KORN J M, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation[J]. Nature Genetics,2008, 40(1):1166-1174.
[6] 郝宏奎,杨立云.同一认定理论地位新探[J].中国人民公安大学学报,2010(4):1-5.
(责任编辑于瑞华)
D918.93
重庆市基础与前沿研究计划项目(cstc2014jcyiA00012)阶段性成果;重庆市教委科学技术项目(KJ1501502)。
庄 艳(1980—),女,新疆昌吉人,博士,讲师。研究方向为植物物证鉴定技术。
