Ce改性Fe2O3催化剂对CO催化氧化的影响
2017-10-20陈然高晓亚王晶陈柯臻钟丽萍雷艳秋梅占强罗永明
陈然,高晓亚,王晶,陈柯臻,钟丽萍,雷艳秋,梅占强,罗永明
Ce改性Fe2O3催化剂对CO催化氧化的影响
陈然,高晓亚,王晶,陈柯臻,钟丽萍,雷艳秋,梅占强,罗永明
(昆明理工大学环境科学与工程学院,云南昆明 650500)
采用共沉淀法制备铁铈氧化物催化剂,考察Ce添加对Fe2O3催化氧化CO性能的影响。活性测试表明,负载Ce后,催化剂活性显著提升,CO的转化率在常温常压下能够达到90%以上,主要得益于添加Ce后的催化剂有着较大的比表面积,更好的氧化还原性能以及在催化剂中形成铁铈固溶体。此外,不同的铁前体对催化剂的活性有较大影响。实验结果表明,以FeCl3作为前体制备出的CeO2/Fe(c)具有最大的比表面积和较好的氧化还原性能,从而显示出最佳的CO氧化活性。
催化;还原;氧化;一氧化碳
一氧化碳(CO)是现今大气污染物主要成分之一,对人体有着很强的毒害作用。CO的来源十分广泛,主要包括煤、石油等燃料的燃烧,大量汽车尾气的排放,冶炼、石化、发电等工厂排放的废气以及一些自然灾害等。空气中CO通过人类呼吸进入体内,把体内氧排挤出去,使得机体缺氧致死;因此探索CO的消除对人类安全和环境保护势在必行。此外,CO的消除在工业生产、军事应用等领域也都有较高的实用价值和应用前景[1-5],出于上述原因,对CO的研究引起了广泛关注。
当今,去除CO最直接、经济、有效的方法是催化氧化法,而此方法的核心在于高效催化剂的设计[6-9]。现今,应用于CO氧化的催化剂种类很多,总体上分为贵金属和非贵金属催化剂两类。贵金属催化剂包括金、银、铂、铑、钯等[10-14],它们有着极佳的催化氧化性能,但是由于贵金属储量少、产量低、价格昂贵,不能大规模广泛应用。而非贵金属催化剂主要包括Co[15-19]、Zn[20]、Ce[21-23]、Cu[24-25]、Mn[26-27]、Ti[28-29]等,他们具有较高的催化氧化性能,同时含量丰富、价格较低,被视为是贵金属催化剂的最佳替代者。
CeO2拥有独特的结构,Ce的周围分散着氧离子,形成立方结构。而且具有较好的储氧能力(OSC)以及氧化还原性能[30]。Ce有Ce3+和Ce4+,相互之间可以发生反应。在氧不足条件下,部分Ce4+被还原为Ce3+,生成含有氧空位的CeO2-x。然而,氧充足时,Ce3+被氧化为Ce4+,致使生成的CeO2-x转化为CeO2,从而使得CeO2拥有较强的储氧性能,因此被大量作为催化剂的载体或者助剂应用于CO去除。但CeO2在制备过程中易高温烧结,在应用于CO低温催化氧化时,其强大的储氧能力会受到严重抑制[31-33],使得CO低温催化氧化的活性很差。而在CeO2的立方萤石结构中引入金属粒子,不仅能提高CeO2的活性,同时也能保持CeO2原有的强储氧能力[34]。
过渡金属元素Fe有+3和+2两种价态,为具备良好的氧化还原能力奠定了坚实的基础。此外,Fe2O3特殊的内在结构有利于增加催化剂的比表面积[35]。铁改性的二氧化铈催化剂与纯的二氧化铈和氧化铁相比较而言,它的氧交换能力有显著提升,而这又归结于铈铁之间的协同作用。KAPTEIJN等[36]报道Fe-Ce氧化物能够形成一种铁铈固溶体,拥有比纯的金属氧化物更好的催化氧化活性。
Fe-Ce氧化物作为催化剂催化氧化CO的研究相对较少,并且在已报道的文献中,CO反应所需的温度大都较高[37-38],而铁前体的选择对Fe-Ce催化剂活性的探究鲜有报道。因此,本文重点探讨Ce改性Fe2O3催化剂对CO氧化性能的差异,并在此基础上,通过优化铁前体选择进一步提高Fe-Ce氧化物催化剂对CO的催化性能。
1 实验部分
1.1 试剂
Ce(NO3)3·6H2O,Fe(NO3)3·9H2O,Fe2(SO4)3,FeCl3,Ce(NO3)3·6H2O,NaOH(0.25mol/L),HCl(0.25mol/L),Na2CO3(0.25mol/L),CO/O2/He(2.0%/1.0%/97.0%,V/V/V),去离子水。实验所用试剂均为分析纯。
1.2 催化剂的制备
采用共沉淀法,首先分别称取一定量的硝酸铁、硫酸铁、氯化铁,再称取准确计算量的硝酸铈。将二者溶于Na2CO3溶液中,然后搅拌2h,同时用0.25mol/L的HCl或者0.25mol/L的NaOH溶液调节混合液pH,在120℃下烘干8h,最后在500℃下焙烧3h。以硝酸铁、硫酸铁、氯化铁作为铁的前体制备出的催化剂分别命名为5%CeO2/Fe(N)、5%CeO2/Fe(S)、5%CeO2/Fe(C)。
1.3 催化剂的活性评价
CO的活性反应在石英玻璃管内进行,在其中装入0.2g催化剂。反应气的体积分数为CO/O2/He(2.0%/1.0%/97.0%)。反应条件为常压,混合气的流速为30mL/min,由质量流量计控制,用气相色谱分析CO转化率。
1.4 催化剂的表征
XRD采用日本理学公司的 D/Max1100型衍射仪对催化剂的结构进行表征。
H2-TPR实验在带有TCD检测器的仪器上进行测试。在石英玻璃管内装入0.1g催化剂,通入Ar/H2(90%/10%),在100℃下吹扫30min,接着从100℃开始,以10℃/min升至900℃。用带有TCD的气相色谱进行测量。
催化剂的比表面积、孔结构等物理性质通过Nova 4200e型吸附仪来测试。测量前,需要对催化剂进行一定的前处理,再置于液氮中进行测量。
2 结果与分析
2.1 Ce添加对Fe2O3催化剂催化氧化CO性能的影响
2.1.1 催化剂的表征
纯Fe2O3、CeO2和5%CeO2/Fe(N)的XRD谱图如图1所示。由图1可知,负载Ce后,催化剂出现明显的CeO2特征峰(2=28.6°,47.6°),表明结晶态CeO2的形成。与纯CeO2相比,峰的衍射强度降低。同时,图中24.2°、33.2°、35.7°、40.9°、49.5°、54.2°等处的衍射峰归属于Fe2O3。添加Ce后,衍射峰的强度较纯Fe2O3相比略微下降,并且衍射角发生偏移,表明部分Ce迁移到Fe2O3结构中,形成铁铈固溶体[39-40]。
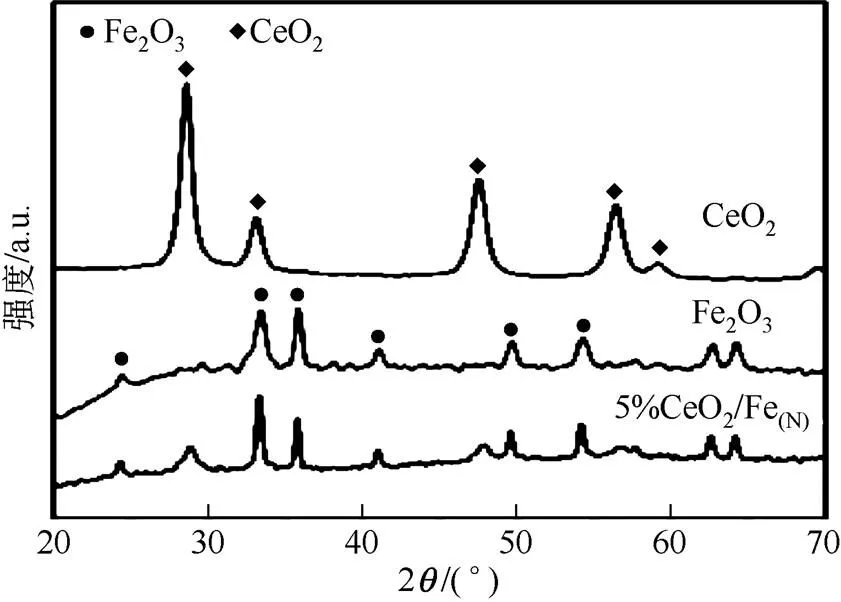
图1 纯Fe2O3、CeO2和5%CeO2/Fe(N)的XRD图
通过N2吸附-脱附法测定的催化剂的比表面积、孔结构等结果如表1。5%CeO2/Fe(N)的比表面积大幅度增加,由于Ce的加入,使得相邻Fe颗粒之间的间距变大[41]。此外,催化剂的孔容较纯Fe2O3增加,孔径减小,由于制备过程中Ce进入Fe2O3的孔结构中,在内表面形成一些结构。

表1 纯Fe2O3和5%CeO2/Fe(N)催化剂的BET测试结果
纯Fe2O3、CeO2和5%CeO2/Fe(N)催化剂的H2-TPR表征见图2。纯Fe2O3在450℃和600℃处显示出两个还原峰。首先是Fe2O3被还原为Fe3O4,接着Fe3O4被还原为Fe0,表明Fe3+和Fe2+存在于催化剂中[42]。纯CeO2的还原峰出现在400℃-表面晶格氧的还原和700℃-体相晶格氧的还原。Ce的添加使催化剂在270℃附近出现了一个还原峰,可能是由于CeO2中表面晶格氧在被还原时,体相中的少量晶格氧过早地扩散到表面被还原,导致峰的出 现[43]。同时,Fe还原峰出现在较低的温度条件下,归结于铁铈固溶体的形成,表明Ce的添加有利于降低还原峰的形成温度,增强氧化还原性[44]。
2.2.2 催化剂的活性评价
催化剂的活性测试如图3所示。由图3可知,5%CeO2/Fe(N)显示出较好的活性,在25℃就能达到90%以上的CO去除率,而纯Fe2O3和CeO2对CO氧化在所示温度范围内几乎没有变化,该结果说明Ce的引入能够大大提升Fe2O3催化氧化CO的 性能。
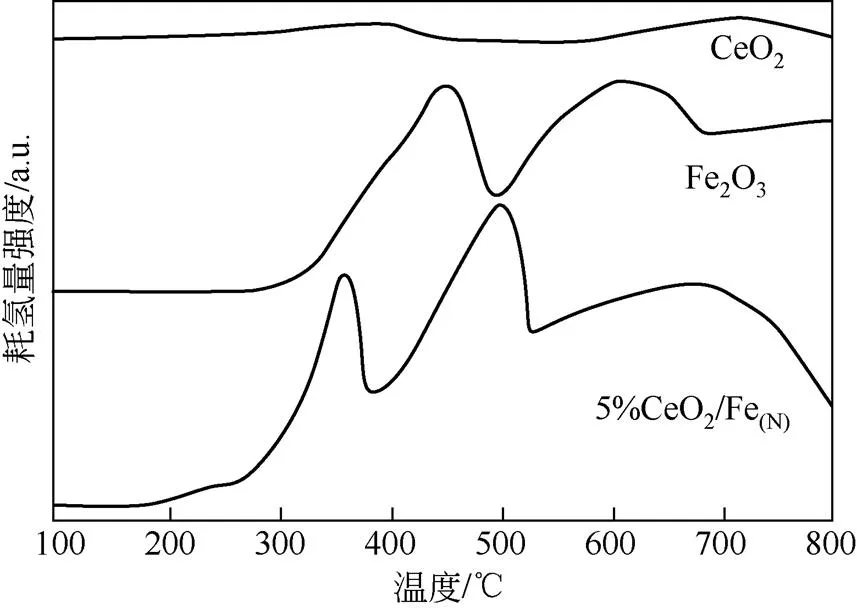
图2 纯Fe2O3、CeO2和5%CeO2/Fe(N)的H2-TPR图
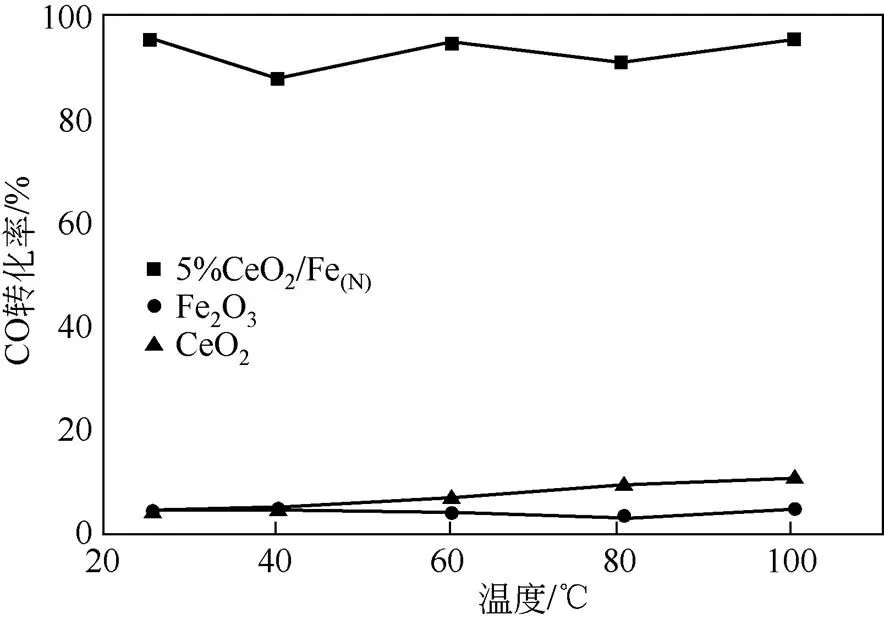
图3 纯Fe2O3、CeO2和5%CeO2/Fe(N)对CO催化氧化的活性图
Ce添加Fe2O3催化剂活性显著提高的原因是:①Ce改性后,催化剂的比表面积增大,使得更多CO附着在催化剂表面;②有较好的氧化还原性能,降低CO氧化所需的温度;③有文献报道,掺杂Ce能够降低Fe2O3形成氧空位的能量[45];④铁铈固溶体的形成能够提升CO氧化性能[44]。
2.2 不同铁前体对CO催化氧化的影响
不同铁盐制备出的Fe-Ce催化剂的XRD图谱见图4。从图中可以看出,CeO2的特征峰出现在28.6°、47.6°处,而24.2°、33.2°、35.7°、40.9°、49.5°、54.2°等处则归属于Fe2O3。由图4可知,铁前体的选择对Fe-Ce氧化物的结构影响较大。图中5%CeO2/Fe(C)基本不显示出Ce的衍射峰,主要是出于Ce较好地分散在Fe表面,同时Fe2O3的衍射峰变弱,由于制备过程中,Fe颗粒变小,导致峰强下降。而对于5%CeO2/Fe(S)和5%CeO2/Fe(N)来说,均能检测出CeO2和Fe2O3的衍射峰。5%CeO2/Fe(N)中的CeO2衍射峰最高,可能由于CeO2颗粒的聚集,增加了CeO2的结晶度。
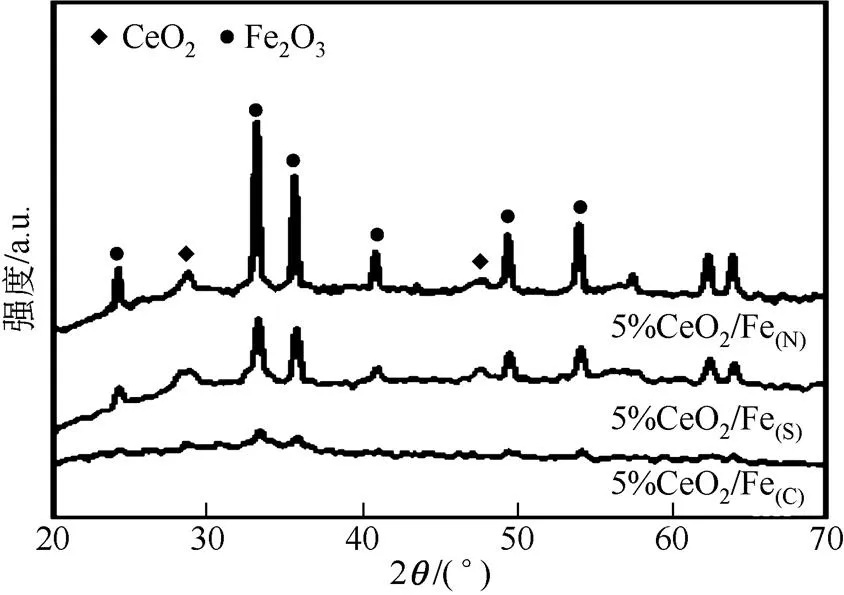
图4 不同铁前体的5%CeO2/Fe催化剂的XRD图
通过N2吸附-脱附法测定的催化剂的比表面积、孔结构等结果如表2。5%CeO2/Fe(C)拥有最大的比表面积,5%CeO2/Fe(N)次之,5%CeO2/Fe(S)最小,较大的比表面积有利于CO附着。5%CeO2/Fe(C)和5%CeO2/Fe(N)有相似的孔容,5%CeO2/Fe(S)最小,较大的孔容有利于更多的Ce进入,有利于铁铈固溶体生成。
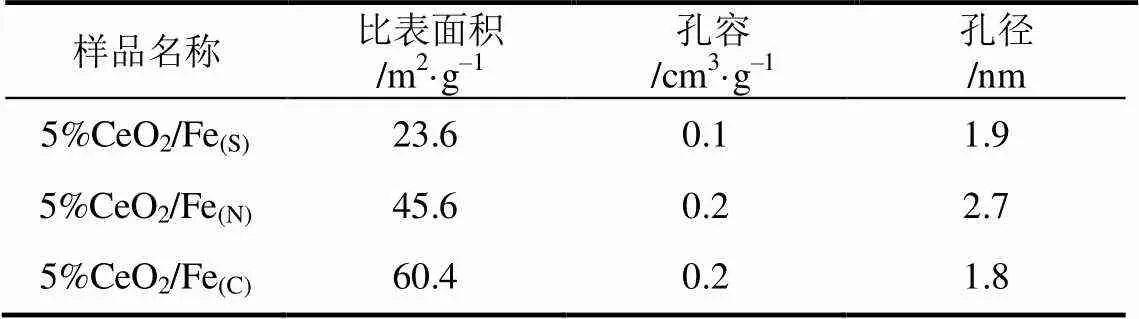
表2 不同铁前体的5%CeO2/Fe催化剂的BET测试结果
不同铁前体制备出的Fe-Ce氧化物的氧化还原性能通过H2-TPR进行测试,其结果见图5。在300℃、400℃和550℃附近均出现3个还原峰,分别命名为、、。峰归属为CeO2中的表面晶格氧在被还原时体相中的少量晶格氧过早地扩散到表面被还原,峰归属为Fe2O3向Fe3O4的还原,则是Fe3O4向Fe0的还原,表明Fe3+和Fe2+两种Fe物种存在于催化剂中[42]。5%CeO2/Fe(S)、5%CeO2/Fe(C)和5%CeO2/Fe(N)催化剂中,,还原峰的出峰情况存在差异,表明5%CeO2/Fe(S),5%CeO2/Fe(C)和5%CeO2/Fe(N)有着不同的氧化还原性能,从而表明不同铁前体与铈之间的相互作用对催化剂的氧化还原性能存在较大影响。5%CeO2/Fe(C)催化剂中的还原峰在260℃()、360℃()和485℃()附近,5%CeO2/Fe(S)催化剂的还原峰出现在310℃()、420℃()和560℃()附近,5%CeO2/Fe(N)的还原峰则出现在270℃(),400℃()和545℃()附近。5%CeO2/Fe(C)催化剂中的、、三个还原峰的温度均低于5%CeO2/Fe(S)和 5%CeO2/Fe(N)催化剂,表明5%CeO2/Fe(C)有着更好的氧化还原性能。而5%CeO2/Fe(S)中、、还原峰温度较高,表明5%CeO2/Fe(S)氧化还原性能较差。
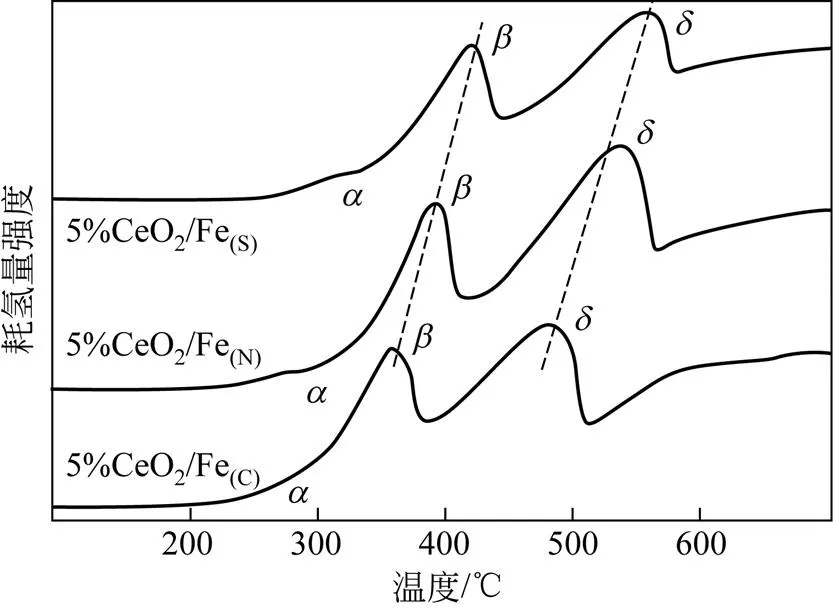
图5 不同铁前体的5%CeO2/Fe催化剂的H2-TPR图
图6为不同铁前体制备的Fe-Ce催化剂催化氧化CO的活性图。由图6可知,铁前体的选择对催化氧化CO的性能影响较大,并且5%CeO2/Fe(C)催化剂有着最佳的催化氧化CO的活性,在25℃下催化氧化CO的转化率达到95%。由图4可知,5%CeO2/Fe(C)中CeO2高度分散在Fe表面,使得Fe与Ce充分接触,增强二者之间的相互作用,由表2可知,5%CeO2/Fe(C)有着最大的比表面积,有益于CO的大量附着,此外由图5可知5%CeO2/Fe(C)还有着较好的氧化还原性能,能够在较低温度将CO氧化。对于5%CeO2/Fe(S)催化剂,一方面,出于未完全分解的硫酸根的存在,覆盖于表面,减少铁铈之间的接触,进而减少二者之间的相互作用。另一方面,根据表2,5%CeO2/Fe(S)的比表面积最低,不利于CO附着,同时TPR结果表明,5%CeO2/Fe(S)的氧化还原性能较差,使得5%CeO2/Fe(S)催化剂催化氧化CO的活性最差。
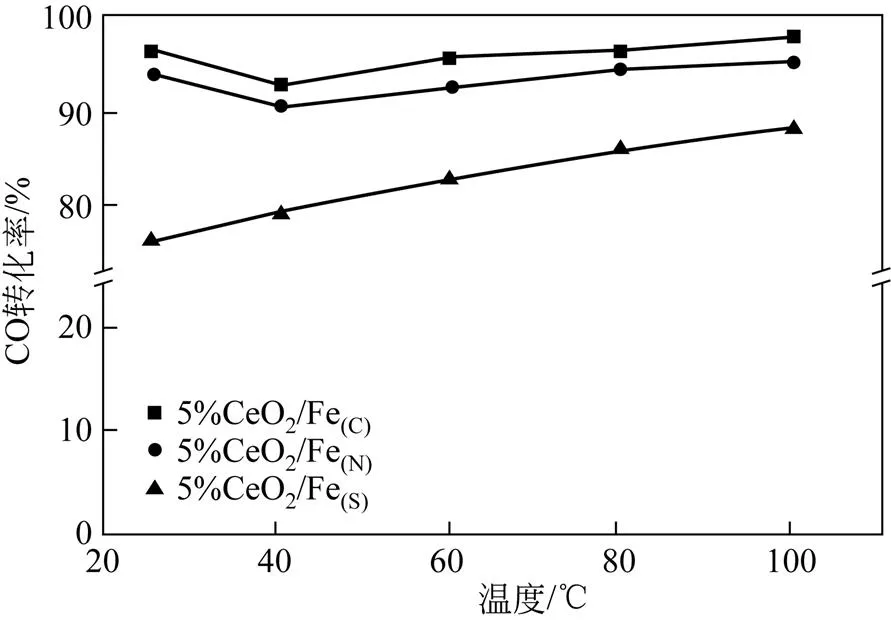
图6 不同铁前体的5%CeO2/Fe催化剂催化氧化CO的活性图
3 结论
(1)Ce改性的Fe-Ce氧化物催化剂大大增加了其催化氧化CO的性能。主要是由于负载Ce后,Fe-Ce的比表面积增大,能够附着更多的CO;同时氧化还原性能增强;而且,将Ce负载Fe2O3上,有利于形成氧空位,此外,还利于催化剂中铁铈固溶体的形成。
(2)不同铁源制备的Fe-Ce氧化物催化剂对CO催化氧化存在较大影响。以氯化铁作为铁源制备出来的催化剂有着最佳的氧化性能。这是由于5%CeO2/Fe(C)拥有着最大的比表面积、较强的铁铈相互作用和较好的氧化还原性能。而以硫酸铁作为铁源制备出的催化剂的活性最差,一方面,出于未完全分解的硫酸根的存在,覆盖于催化剂表面,减少铁铈之间相互作用。另一方面,5%CeO2/Fe(S)的比表面积最低,不利于CO的附着,同时H2-TPR结果表明,5%CeO2/Fe(S)的氧化还原性能较差,导致5%CeO2/Fe(S)有着最差的催化氧化活性。因此,5%CeO2/Fe(C)对催化氧化CO有着最佳的活性。
[1] 王东辉,程代云,郝正平,等. 纳米金催化剂上CO低(常)温氧化的研究[J]. 化学进展,2002,14(5):360-367.
WANG D H,CHEN D Y,HAO Z P,et al. Carbon monoxide low-temperature oxidation over nanosize gold catalyst[J]. Progress in Chemistry,2002,14(5):360-367.
[2] FUNAZAKI N,HEMMI A,ITO S,et al. Development of carbon monoxide detector using Au fine particles doped α-Fe2O3[J]. Sensors and Actuators B,1993,13/14: 536-538.
[3] 张俊,陈婧,黄新松,等. CO催化氧化用纳米材料及其最新研究成果[J]. 化学进展,2012,24:1245-1251.
ZHANG J,CHEN J,HUANG X S,et al. Recent research progress and applications of nano catalytic materials for CO oxidation[J]. Progress in Chemistry,2012,24:1245-1251.
[4] YUAN Y,KOZLVOA A P,ASAKURA K,et al. Supported Au catalysts prepared from Au phosphine complexes and As-precipitated metal hydroxides:characterization and low-temperature CO oxidation[J]. Journal of Catalysis,1997,170:191-199.
[5] CHEN G X,LI Q L,WEI Y C,et al. Low temperature CO oxidation on Ni-promoted CuO-CeO2catalysts[J]. Chinese Journal of Catalysis,2013,34:322-329.
[6] XIE X,LI Y,LIU Z Q,et al. Low-temperature oxidation of CO catalysed by Co3O4nanorods[J]. Nature,2009,458:746-749.
[7] ZHONG L S,HU J S,CAO A M,et al. 3D flowerlike ceria micro/nanocomposite structure and its application for water treatment and CO removal[J]. Chemistry of Material,2007,19:1648-1655.
[8] LIU X,LIU M H,LUO Y C,et al. Strong metal-support interactions between gold nanoparticles and ZnO nanorods in CO oxidation[J]. Journal of the American Chemical Society,2012,134:10251-10258.
[9] LIN H K,CHIU H C,TSAI H C,et al. Characterization and catalytic oxidation of carbon monoxide over cobalt oxide[J]. Catalysis Letters,2003,88:169-174.
[10] 刘东亮,刘道胜,张晓彤,等. CO低温氧化负载金催化剂研究进展[J]. 化工进展,2007,26(8):1110-1115.
LIU D L,LIU D S,ZHANG X T,et al. Studies on low-temperature oxidation of CO by Au cataIyst[J]. Chemical Industry and Engineering Progress,2007,26(8):1110-1115.
[11] 鲁继青,罗孟飞,辛勤. 纳米金催化剂在 CO 低温氧化和选择性氧化中的研究进展[J]. 化工进展,2007,26(3):306-309.
LU J Q,LUO M F,XIN Q. Nano-sized gold catalysts for CO low-temperature oxidation and selective oxidation[J]. Chemical Industry and Engineering Progress,2007,26(3):306-309.
[12] KIM Y,SHI S K,WHITE J M. Oxygen inhibition of CO oxidation on polycrystalline Rh[J]. Journal of Catalysis,1980,61:374-377.
[13] YAO Y F Y. The oxidation of CO and hydrocarbons over noble metal catalysts[J]. Journal of Catalysis,1984,87:152-162.
[14] SCHWANKNER R J,EISWIRTH M,MÖLLER P,et al. Kinetic oscillations in the catalytic CO oxidation on Pt(100):periodic perturbations[J]. Journal of Chemical Physics,1985,83(4):1578-1587.
[15] HU L,SUN K,PENG Q,et al. Surface active sites on Co3O4nanobelt and nanocube model catalysts for CO oxidation[J]. Nano Research,2010,3:363-368.
[16] 陈霞,张俊丰,黄妍,等. CuCoO/TiO2催化氧化CO性能[J]. 化工进展,2009,28(8):1355-1359.
CHEN X,ZHANG J F,HUANG Y,et al. Catalytic performance of CuCoO/TiO2for oxidation of carbon monoxide[J]. Chemical Industry and Engineering Progress,2009,28(8):1355-1359.
[17] JIA C J,SCHWICKARDI M,WEIDENTHALER C,et al. Co3O4-SiO2nanocomposite: a very active catalyst for CO oxidation with unusual catalytic behavior[J]. Journal of the American Chemical Society,2011,133:11279-11288.
[18] ROYER S,DUPREZ D. Catalytic oxidation of carbon monoxide over transition metal oxides[J]. ChemCatChem,2011,3:24-65.
[19] DENG S,XIAO X,XING X,et al. Structure and catalytic activity of 3D macro/mesoporous Co3O4for CO oxidation prepared by a facile self-sustained decomposition of metal-organic complexes[J]. Journal of Molecular Catalysis A:Chemical,2015,398:79-85.
[20] WANG G Y,ZHANG W X,LIAN H L,et al.Effect of calcination temperatures and precipitant on the catalytic performance of Au/ZnO catalysts for CO oxidation at ambient temperature and in humid circumstances[J]. Applied Catalysis A:General,2003,239:1-10.
[21] YAKIMOVA M S,IVANOV V K,POLEZHAEVA O S,et al. Oxidation of CO on nanocrystalline ceria promoted by transition metal oxides[J]. Doklady Chemistry,2009,427:186-189.
[22] ZOU Q Z,MENG M,LI Q,et al. Surfactants-assisted synthesis and characterizations of multicomponent mesoporous materials Co-Ce-Zr-O and Pd/Co-Ce-Zr-O used for low-temperature CO oxidation[J]. Materials Chemistry and Physics,2008,109:373-380.
[23] SASIKALA R,GUPTA N M,KULSHRESHTHA S K.Temperature-programmed reduction and CO oxidation studies over Ce-Sn mixed oxides[J]. Catalysis Letters,2001,71:69-73.
[24] 士丽勄,郑德山,王素勄. 富氢气氛中CO优先氧化CuO-CeO2基催化剂的研究进展[J]. 化工进展,2011,30(9):1956-1960.
SHI L M,ZHENG D S,WANG S M. Research progress of CuO-CeO2-based catalysts for preferential oxidation of CO in hydrogen-rich gas[J]. Chemical Industry and Engineering Progress,2011,30(9):1956-1960.
[25] 张文丽,刘娜,丁素萍,等. 逆CeO2-CuO催化剂用于优先氧化CO的性能[J]. 化工进展,2011,30(8):1744-1748.
ZHANG W L,LIU N,DING S P,et al. Study on catalytic performance of CeO2/CuO catalyst for preferential CO oxidation[J]. Chemical Industry and Engineering Progress,2011,30(8):1744-1748.
[26] LIANG S,TENG F,BULGAN G,et al. Effect of phase structure of MnO2nanorod catalyst on the activity for CO oxidation[J].Journal of Physical Chemistry C,2008,112:5307-5315.
[27] XU R,WANG D,WANG X,et al. Surface structure effects in nanocrystal MnO2and Ag/MnO2catalytic oxidation of CO[J].Journal of Catalysis,2006,237:426-430.
[28] YAN W F,CHEN B,MAHURIN S M,et al.Preparation and comparison of supported gold nanocatalysts on anatase,brookite,rutile,and P25 polymorphs of TiO2for catalytic oxidation of CO[J]. Journal of Physical Chemistry B,2005,109:10676-85.
[29] GREEN I X Y,TANG W J,NEUROCK M,et al.Spectroscopic observation of dual catalytic sites during oxidation of CO on a Au/TiO2catalyst[J]. Science,2011,333:736-739.
[30] LI K,ZHOU W,WANG X,et al. CeO2based oxygen storage compounds II.Adding precious metal to improve oxygen storage capacity[J]. Journal of the Chinese Rare Earth Society,2004,22:32-34.
[31] TROVARELLI A,LEITENBURG C D,DOLCETTI G,et al.Design better cerium-based oxidation catalysts[J]. Chemtech,1997,27:32-37.
[32] DESHPANDE P A,ARUNA S T,MADRAS G. CO oxidation by CeO2-Al2O3-CeAlO3hybrid oxides[J]. Catalysis Science and Technology,2011,1:1683-1691.
[33] KUBSH J E,RIECK J S,SPENCER N D. Cerium oxide stabilization: physical property and three-way activity considerations[J]. Studies in Surface Science and Catalysis,1991,71:125-138.
[34] TROVARELLI A. Catalytic properties of ceria and CeO2-containing materials[J]. Catalysis Reviews,1996,38(4):439-520.
[35] 赵志利,李建伟,陈标华.三氧化二铁晶型对铁铬系高温变换催化剂性能的影响[J]. 现代化工,2006,26(2):143-146.
ZHAO Z L,LI J W,CHEN B H. Effect of Fe2O3crystal structure on the performance of Fe2O3/Cr2O3high temperature water gas shift catalyst[J]. Modern Chemical Industry,2006,26(2):143-146.
[36] KAPTEIJN F,LOPEZ G M,MELIANCABRERA I,et al. Synergy of FeCe1−xO2mixed oxides for N2O decomposition[J]. Journal of Catalysis,2006,239:340-346.
[37] LIU X,LIU J,CHANG Z,et al. Crystal plane effect of Fe2O3with various morphologies on CO catalytic oxidation[J]. Catalysis Communication,2011,12:530-534.
[38] ZHONG Z Y,HO J,TEO J,et al. Synthesis of porous α-Fe2O3nanorods and deposition of very small gold particles in the pores for catalytic oxidation of CO[J]. Chemistry of Materials,2007,19:69-73.
[39] 孙令玥. 铈铁氧载体化学链储氢-制氢性能研究[D]. 昆明:昆明理工大学,2015.
SUN L Y. Hydrogen storage of cerium iron oxide carrier chain of chemical-research on hydrogen production performance[D]. Kunming:Kunming University of Science and Technology,2015.
[40] LI X M,HAN D Z,WANG H,et al. Propene oligomerization to high-quality liquid fuels over Ni/HZSM-5[J]. Fuel,2015,144:9-14.
[41] TANG L,YAMAGUCHI D,BURKE N,et al. Methane decomposition over ceria modified iron catalysts[J]. Catalysis Communications,2010,11:1215-1219.
[42] ZIELINSKI J,ZGLINICKA I,ZNAK L,et al. Reduction of Fe2O3with hydrogen[J]. Applied Catalysis A:General,2010,381:191-196.
[43] ZHU X,WEI Y,WANG H,et al. Ce-Fe oxygen carriers for chemical-looping stream methane reforming[J]. International Journal of Hydrogen Energy,2013,38:4492-4501.
[44] QIAO D S,LU G Z,LIU X H,et al. Preparation of Ce12xFeO2solid solution and its catalytic performance for oxidation of CH4and CO[J]. Journal of Material Science,2011,46:3500-3506.
[45] 刘雄姗. Fe-CeO2(111)表面催化活性及CO吸附与氧化第一性原理研究[D]. 大连:大连理工大学,2015.
LIONG X S. First-principles study of the catalytic activities of Fe-CeO2(111) surface and CO adsorption and oxidation at the doped surface[D]. Dalian:Dalian University of Technology,2015.
Effect of Ce addition on Fe2O3catalyst towards CO catalytic oxidation
CHEN Ran,GAO Xiaoya,WANG Jing,CHEN Kezhen,ZHONG Liping,LEI Yanqiu,MEI Zhanqiang,LUO Yongming
(Faculty of Environmental Science and Engineering,Kunming University of Science and Technology,Kunming 650500,Yunnan,China)
A series of iron-ceria oxide catalysts were prepared by-precipitation method and the effects of Ce adding into Fe2O3on CO catalytic oxidation activity were investigated. Activity result shows that the addition of Ce increases the CO catalytic activity significantly,which can attain more than 90% CO conversion at ambient temperature and atomospheric pressure. Higher surface area and better redox ability than those of pure Fe2O3,and the formation of iron-ceria solid solution in the catalyst may be responsible for the increased catalytic activity. Additionally,different iron precursor has great influence on the CO catalytic oxidation. Sample that was prepared by using FeCl3asiron precursor shows the best catalytic performance.
catalysis;reduction;oxidation;carbon monoxide
X511
A
1000–6613(2017)10–3737–06
10.16085/j.issn.1000-6613.2017-0090
2017-01-16;
2017-03-29。
国家自然科学基金项目(U1402233, 21267011,21367015)。
陈然(1990—),男,硕士研究生,主要从事大气污染的防治研究。
罗永明,教授,博士生导师,主要从事先进环境功能材料的制备及其在污染防治与污染物资源化利用研究。E-mail:environcatalysis222@yahoo.com。
