氟丙嘧草酯的合成与生物活性
2017-10-16郭正峰英君伍杨辉斌
郭正峰,陈 霖,英君伍,杨辉斌,李 斌*
(1.沈阳化工研究院有限公司,沈阳 110021;2.沈阳中化农药化工研发有限公司,新农药创制与开发国家重点实验室,沈阳 110021)
◆研究与开发◆
氟丙嘧草酯的合成与生物活性
郭正峰1,2,陈 霖2,英君伍2,杨辉斌2,李 斌2*
(1.沈阳化工研究院有限公司,沈阳 110021;2.沈阳中化农药化工研发有限公司,新农药创制与开发国家重点实验室,沈阳 110021)
氟丙嘧草酯是先正达公司开发的非选择性脲嘧啶类除草剂,2001年上市。以5-氨基-2-氯苯甲酸为起始原料,经酰氯化并与2-羟基异丁酸烯丙酯的钠盐发生酯化反应,得到5-氨基-2-氯苯甲酸(1-甲基-1-烯丙氧羰基乙)酯(M-3);该中间体与3-(3,3-二甲基脲基)-4,4,4-三氟巴豆酸乙酯进行环合后,进一步经N-甲基化反应得到目标产物。目标化合物和部分中间体的结构经1H NMR和LC-MS确证。苗后除草活性测试结果表明,氟丙嘧草酯对百日草和苘麻具有优异的除草活性,在9.4 g/hm2有效成分用量下对二者的防除效果分别为100%和65%。
除草剂;氟丙嘧草酯;合成;除草活性
Abstract:Butafenacil is a kind of non-selective uracil herbicide developed by Syngenta and was marketed in 2001.Based on the prior literatures,the intermediate 5-amino-2-chlorobenzoic acid(1-methyl-1-allyloxycarbonylethyl)ester(M-3)was synthesized from 5-amino-2-chlorobenzoic acid by acylation reaction and esterified with the sodium salt of allyl 2-hydroxyisobutyrate.M-3 reacted with 3-(3,3-dimethylureido)-4,4,4-trifluorocrotonic acid ethyl ester,then the N-methylation reaction was carried out to obtain butafenacil.The structures of the target compound and some intermediates were confirmed by1H NMR and LC-MS.The results of herbicidal activity at greenhouse showed that butafenacil had excellent herbicidal activities against Zinnia elegans and Abutilon theophrasti.At the active ingredient dosage of 9.4 g/hm2,the herbicidal activities on Zinnia elegans and Abutilon theophrasti were 100%and 65%.
Key words:herbicide;butafenacil;synthesis;herbicidal activity
氟丙嘧草酯(英文通用名butafenacil,商品名Inspire)是先正达公司开发的一种非选择性脲嘧啶类除草剂[1-4]。它于2001年上市,主要用于葡萄园、棉花及非耕地防除禾本科杂草、阔叶杂草和莎草等,苗前和苗后除草用量均为75~150 g/hm2。
氟丙嘧草酯,CAS号134605-64-4,分子式C20H18ClF3N2O6,相对分子质量474.82。化学名称:2-氯-5-[1,2,3,6-四氢-3-甲基-2,6-二氧-4-(三氟甲基)嘧啶-1-基]苯甲酸[1-(烯丙氧基羰基)-1-甲基乙]酯,化学结构式如图1所示。
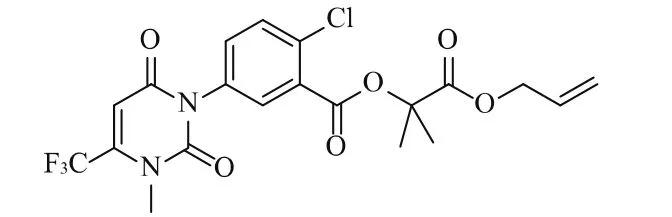
图1 氟丙嘧草酯化学结构式
1 合成路线
氟丙嘧草酯文献报道的合成路线主要有如下2种。
路线1[1]:
以5-硝基-2-氯苯甲酸为起始原料,经酯化、还原、胺解、缩合、环合、甲基化、加氢还原、酰氯化、缩合等9步反应制得目标物(见图2)。该合成路线首先对活性基团羧基进行保护,经一系列反应后再用催化加氢的方法将羧基脱保护,最后与2-羟基异丁酸烯丙酯缩合生成目标物。
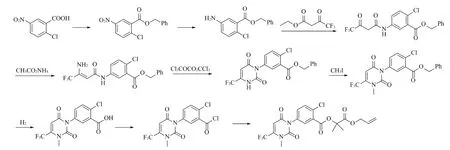
图2 氟丙嘧草酯合成路线1
路线2[2]:
路线2也以5-硝基-2-氯苯甲酸为起始原料,经酯化、还原、异氰酸化、环合等4步反应制得目标产物氟丙嘧草酯,合成路线见图3。与路线1相比,该合成路线利用羧酸与2-羟基异丁酸烯丙酯直接进行酯化反应,省去了羧基的保护和脱保护2步反应,最后与3-甲氨基-4,4,4-三氟巴豆酸乙酯直接环合即可制得目标物。
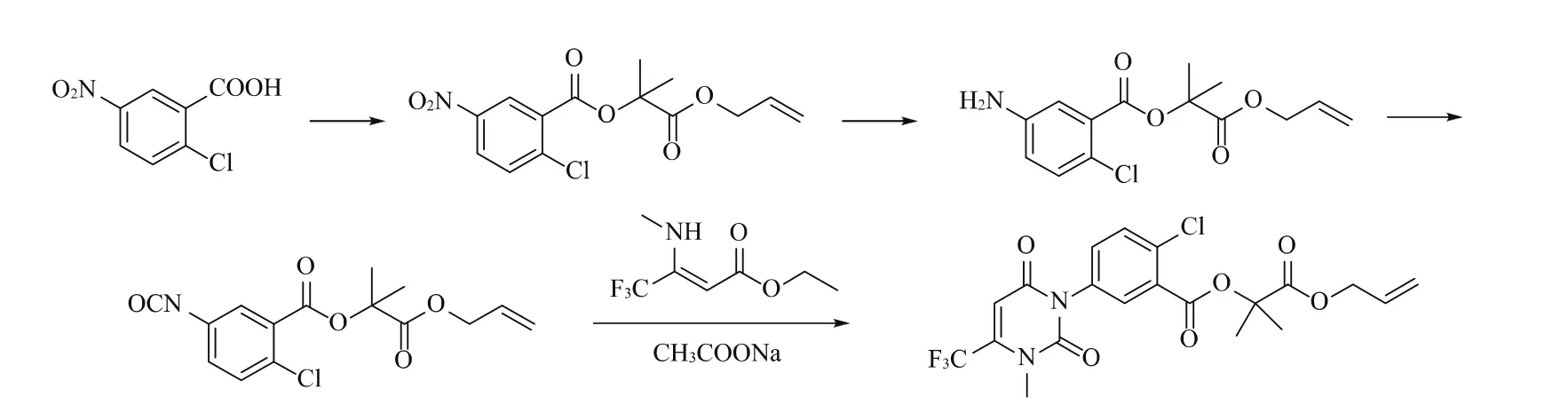
图3 氟丙嘧草酯合成路线2
本研究在现有文献基础上,综合考虑原料成本、实验操作、反应条件等方面内容,以5-氨基-2-氯苯甲酸为起始原料,与氯化亚砜反应制得酰氯(M-1),再与2-羟基异丁酸烯丙酯的钠盐(M-2)发生酯化反应得到5-氨基-2-氯苯甲酸(1-甲基-1-烯丙氧羰基乙)酯(M-3)。另将3-氨基-4,4,4-三氟巴豆酸乙酯与N,N-二甲氨基甲酰氯反应制成3-(3,3-二甲基脲基)-4,4,4-三氟巴豆酸乙酯(M-4)后,与M-3在酸性条件下环合得到M-5,进一步与碘甲烷发生N-甲基化得到目标物。该反应路线操作简便,反应条件温和,适宜实验室操作。合成路线如图4所示。
在中间体M-3的合成过程中,利用了异硫氰酸酯易水解放出二氧化硫从而还原为氨基的特点,成功合成中间体M-3。与文献合成路线2硝基化合物还原为氨基化合物相比,简化了操作步骤。产品结构经核磁氢谱、质谱验证。由核磁数据可知:嘧啶5位氢为单峰,化学位移为6.38;亚甲基2个氢为双重峰,化学位移为4.65,耦合常数为5.4 Hz;嘧啶3位的甲基3个氢为单峰,化学位移3.56;2个甲基6个氢为单峰,化学位移为1.70。
2 实验部分
2.1 仪器与试剂
主要试剂:所用试剂为市售化学纯或分析纯。
主要仪器:Mercury 600(Varian)核磁共振仪(CDCl3为溶剂,TMS为内标);天津分析仪器厂生产的RY-1型熔点仪;Agilent 1100系列高效液相色谱仪;LC-MSD-Trap-VL型液质联用仪。

图4 本文合成路线
2.2 中间体M-1的合成
向250 mL反应瓶中,依次加入5-氨基-2-氯苯甲酸10.08 g(50.00 mmol)、甲苯70 mL,室温搅拌5 min后逐滴加入氯化亚砜23.79 g(200 mmol),滴加完毕后,加热回流至无氯化氢气体放出为止。减压蒸除甲苯和多余的氯化亚砜,降温冷却,得到黄色油状物,加入50 mL无水四氢呋喃,搅拌溶解,密封。
2.3 中间体M-2的合成
冰水浴下,向500 mL反应瓶中加入120 mL无水四氢呋喃,降温30 min后分批加入60%氢化钠2.00 g(50 mmol),搅拌10 min后逐滴加入2-羟基异丁酸烯丙酯5.77 g(40.00 mmol),滴加完毕后,室温搅拌4 h后停止反应。
2.4 中间体M-3的合成
将上述M-1的四氢呋喃溶液逐滴加入到M-2溶液中,反应体系逐渐由白色变为黄色,滴加完毕后室温搅拌2 h,TLC显示反应无原料剩余。
将混合物倒入100 mL乙醇中,搅拌30 min后减压蒸除溶剂,向剩余物中加入100 mL水,用乙酸乙酯(100 mL×2)萃取,合并有机相。有机相用水洗,饱和食盐水洗,无水硫酸镁干燥,减压脱溶,柱色谱提纯得到2.52 g黄色油状物M-3,质量分数86%,收率18%(以2-羟基异丁酸烯丙酯计)。黄色油状物直接用于下一步反应。
1H NMR (600 MHz,CDCl3) δ:7.17(d,J=9.0 Hz,1H,Ph-H),7.08(d,J=3.0 Hz,1H,Ph-H),6.70(dd,J1=2.4 Hz,J2=8.4 Hz,1H,Ph-H),5.95-5.88(m,1H,-CH2-CH=),5.33(dq,J1=1.2 Hz,J2=2.4 Hz,J3=16.8 Hz,1H,=CH2),5.23 (dq,J1=1.2 Hz,J2=2.4 Hz,J3=12 Hz,1H,=CH2),4.66(dt,J1=1.2 Hz,J2=6.0 Hz,2H,OCH2),3.77(brs,2H,NH2),1.70(s,6H,C(CH3)2)。
LC-MS(m/z):C14H16ClNO4,297.08(计算值);[M+Na]+,320.0(实验值)。
2.5 中间体M-4的合成
向500 mL反应瓶中加入3-氨基-4,4,4-三氟巴豆酸乙酯9.16 g(50.00 mmol)和 150 mL无水四氢呋喃,冰水浴下向上述溶液中分批加入氢化钠6.00 g(150 mmol)。保温反应1 h后,逐滴加入N,N-二甲氨基甲酰氯,滴加完毕后加热回流1 h,TLC显示无原料剩余。然后将混合物缓慢倒入乙醇中,室温搅拌10 min后,减压脱溶,剩余物经柱色谱提纯得6.2 g红色油状物M-4,粗收率49%,直接用于下一步反应。
2.6 中间体M-5的合成[4]
向100 mL反应瓶中加入1.49 g(5.00 mmol)M-3、1.90 g(7.50 mmol)M-4和30 mL冰醋酸,加热回流4 h后TLC显示反应完全。
减压蒸除乙酸后,将剩余物倒入20 mL水中,然后加入乙酸乙酯(30 mL×2)萃取,合并有机相。有机相用水洗,饱和食盐水洗,无水硫酸镁干燥,减压脱溶,经过柱色谱提纯得到1.21 g白色固体,质量分数98.5%,收率51.7%,熔点176~178℃。
1H NMR(600 MHz,CDCl3)δ:9.57(brs,1H,NH),7.74(d,J=2.4 Hz,1H,Ph-H),7.59 (d,J=8.4 Hz,1H,Ph-H),7.30(dd,J1=3.0 Hz,J2=8.4 Hz,1H,Ph-H),6.25 (s,1H,Pyrimidine-H),5.94-5.87(m,1H,-CH2-CH=),5.32(dd,J1=1.2 Hz,J2=17.4 Hz,1H,=CH2),5.24(dd,J1=1.2 Hz,J2=10.8 Hz,1H,=CH2),4.66(d,2H,J=5.4 Hz,OCH2),1.70(s,6H,C(CH3)2)。
LC-MS(m/z):C19H16ClF3N2O6,460.06(计算值);[M+Na]+,483.1(实验值)。
2.7 氟丙嘧草酯的合成
向100 mL反应瓶中加入1.21 g(2.63 mmol)M-5、20 mL DMF,室温搅拌5 min后加入碳酸钾0.44 g(3.15 mmol),继续室温搅拌10 min后加入碘甲烷0.50 g(3.15 mmol),室温搅拌2 h后TLC显示反应完全。
将混合物倒入40 mL水中,加入乙酸乙酯(30 mL×2)萃取,合并有机相。有机相用水洗,饱和食盐水洗,无水硫酸镁干燥,减压脱溶,柱色谱提纯得到0.62 g粗产品。用乙酸乙酯和石油醚进行重结晶,得到0.54 g白色固体,质量分数99.2%,收率43%,熔点111~112℃(文献值113℃[2])。
1H NMR (600 MHz,CDCl3) δ:7.73(d,J=3.0 Hz,1H,Ph-H),7.59(d,J=8.4 Hz,1H,Ph-H),7.29(dd,J1=3.0 Hz,J2=8.4 Hz,1H,Ph-H),6.38(s,1H,Pyrimidine-H),5.94-5.87(m,1H,-CH2-CH=),5.32 (dd,J1=1.2 Hz,J2=17.4 Hz,1H,=CH2),5.22(dd,J1=1.2 Hz,J2=10.8 Hz,1H,=CH2),4.65 (d,J=5.4 Hz,2H,OCH2),3.56(s,3H,N-CH3),1.70(s,6H,C(CH3)2)。
LC-MS(m/z):C20H18ClF3N2O6,474.08(计算值);[M+Na]+,497.1(实验值)。
3 除草活性测定
将定量的阔叶杂草(百日草和苘麻)种子分别播于直径7 cm装有营养土的纸杯中,播后覆土1 cm,镇压,淋水后在室温条件下按常规方法培养。阔叶杂草长至2~4叶期,按照试验设计剂量,在履带式作物喷雾机(英国Engineer Research Ltd.)上进行茎叶喷雾处理,喷雾压力1.95 kg/cm2,喷雾量500 L/hm2,履带速度1.48 km/h。试验设3次重复。试材处理后置于操作大厅,待药液自然阴干后,放于温室内按常规方法管理,处理后定期目测调查供试药剂对杂草的防除效果。
氟丙嘧草酯对百日草、苘麻的除草活性见表1。
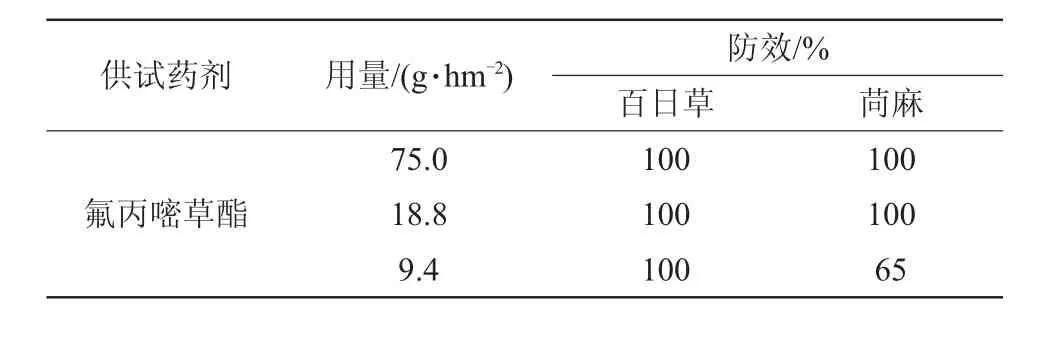
表1 氟丙嘧草酯生物活性结果
4 结果与讨论
本文探索了氟丙嘧草酯的一种新的合成途径。该方法以5-氨基-2-氯苯甲酸为起始原料,经酰氯化并与2-羟基异丁酸烯丙酯的钠盐发生酯化反应,得到5-氨基-2-氯苯甲酸(1-甲基-1-烯丙氧羰基乙)酯(M-3);M-3与3-(3,3-二甲基脲基)-4,4,4-三氟巴豆酸乙酯(M-4)进行环合后,进一步经N-甲基化反应得到目标产物。目标化合物结构通过核磁氢谱、质谱验证。此路线具有原料成本低廉,操作简便,反应条件温和等特点,适宜在实验室中操作。在中间体M-3的合成过程中,利用了异硫氰酸酯易水解放出二氧化硫从而还原为氨基的特点,成功合成中间体M-3。与文献合成路线2硝基化合物还原为氨基化合物相比,简化了操作步骤。但目前各步反应收率明显偏低,有待进一步研究。文中还对目标化合物进行了生物活性测定。结果表明,该化合物对百日草及苘麻具有优异的防除效果,在9.4 g/hm2有效成分用量下,对苘麻的防除效果达到65%,对百日草的防除效果达到100%。
[1]李斌,杨华铮,刘斌,等.用作除草剂的1-嘧啶酮基-4-氯-5苯甲酸酯类化合物及其制备方法:ZL,200510013324.X[P].2005-10-26.
[2]Sting A R.Process for the Production of 3-Aryl-uracils:EP,0831091[P].1998-03-25.
[3]Sting A R,Siegrist U,Studer M,et al.Preparation of 3-Aryl-uracil Derivative Useful as Herbicide:DE,19741411[P].1998-03-26.
[4]Kameswaran V.Process for the Preparation of 6-(Perfluoroalkyl)-uracil Compounds from Urea Compounds:WO,0049004[P].2000-08-24.
(责任编辑:柏亚罗)
Synthesis and Bioactivity of Butafenacil
GUO Zheng-feng1,2,CHEN Lin2,YING Jun-wu2,YANG Hui-bin2,LI Bin2*
(1.Shenyang Chemical Industry Research Institute Co.,Ltd.,Shenyang 110021,China;2.State Key Laboratory of the Discovery and Development of Novel Pesticide,Shenyang Sinochem Agrochemicals Research and Development Co.,Ltd.,Shenyang 110021,China)
TQ 457.2+3
A
10.3969/j.issn.1671-5284.2017.05.004
2017-05-12
郭正峰(1990—),男,辽宁省丹东市人,硕士研究生。研究方向:新农药创制。E-mail:18842350864@163.com
李斌(1964—),男,教授。研究方向:新农药创制。E-mail:libin1@sinochem.com
