我国近岸多室草苔虫(Bugula neritina)的群体遗传分化研究
2017-10-09李海刘巧红唐雪颖陈武个丁少雄
李海,刘巧红,唐雪颖,陈武个,丁少雄*
(1. 国家海洋局第三海洋研究所 海洋生物与生态实验室,福建 厦门 361005;2. 福建省海洋生物资源开发利用协同创新中心,福建 厦门 361102;3.厦门海洋职业技术学院,福建 厦门 361012)
我国近岸多室草苔虫(Bugula neritina)的群体遗传分化研究
李海1,2,刘巧红2,唐雪颖2,陈武个3,丁少雄2*
(1. 国家海洋局第三海洋研究所 海洋生物与生态实验室,福建 厦门 361005;2. 福建省海洋生物资源开发利用协同创新中心,福建 厦门 361102;3.厦门海洋职业技术学院,福建 厦门 361012)
基于线粒体控制区序列和SLAF-seq,对重要药源生物多室草苔虫的群体遗传分化水平开展了研究。控制区序列中检测到8个单倍型,单倍型多样性(h)和核苷酸多样性(π)分别为0.130 7和0.000 7,单倍型网络图和NJ系统进化树的结构都较简单,无明显拓扑结构。中性检验和核苷酸不配对分析结果均表明多室草苔虫未经历过大规模群体扩张。Fst和AMOVA分析显示遗传变异主要来自于群体内。SLAF建库共开发得到214 409个SLAF标签,其中多态性SLAF标签23 437个,共开发出99 432个SNP位点。群体间的遗传距离较小,且低于群体内的遗传距离。基于SNP所做的系统发育树和群体遗传结构分析表明,各群体之间没有显著的遗传结构。综上所述,我国沿海多室草苔虫的遗传多样性水平较低,不同地理群体之间不存在显著的遗传结构。多室草苔虫较强的扩散能力是造成上述结果的主要原因。另外,本研究还验证和讨论了SLAF-seq应用在海洋生物群体遗传分化研究中的可行性和优势。
多室草苔虫;群体遗传分化;线粒体DNA;SLAF-seq;简化基因组
1 引言
多室草苔虫(Bugulaneritina),又称总合草苔虫,隶属于苔藓动物门(Bryozoa),裸唇纲(Cymndaemata),唇口目(Cheilostomida),草苔虫科(Bugulidae),草苔虫属(Bugula),系环热带广布种,广泛地分布于世界各地的中低纬海域[1]。在中国范围内的表层水体中,多室草苔虫只分布于东海和南海等南方海区[2]。作为一类常见的污损苔虫,多室草苔虫经常附着于船底、浮标、石油平台等人造水下设施以及养殖网箱、定置渔网等水产养殖设施上。
多室草苔虫还是一种重要的海洋药源生物。到目前为止,国内外学者一共从多室草苔虫中成功分离到22个抗癌活性成分[3-5]。草苔虫内酯bryostatin系列成分,具有抗肿瘤和免疫调节作用[6],其中,bryostatin 1对于阿尔茨海默症和许多癌症更有着显著的疗效,是草苔虫内酯中最有希望成药的抗癌药物来源[7-11]。目前,该活性成分已经被美国食品与药品管理局批准进入临床 Ⅱ 期试验[12]。然而,bryostatin 1只在美国的东太平洋沿岸等海域生长的多室草苔虫中发现[4],从中国沿海生长的多室草苔虫中却一直未能成功分离。类似的由于多室草苔虫生长地点的地域性差别而造成活性产物不同的现象在世界其他地方也同样存在[4,13],在中国范围内,从生长于不同地方的多室草苔虫中提取得到的草苔虫内酯亦存在一定差异[4],但其影响因素是来源于内在的遗传差异还是外在的环境因子目前尚不得知。而关于这种重要药源生物的生物地理学和群体遗传学的研究至今仍少有报道。因此,开展中国沿海多室草苔虫的群体遗传分化研究不仅可为类似污损生物的扩散方式研究提供借鉴,亦可为该重要药源生物的种质资源保护及其活性产物研发提供必要参考。
线粒体DNA(mtDNA)已被众多学者证实其在海洋生物群体遗传学研究中的适用性[14-17]。而SLAF-seq作为一种基于限制性酶切的简化基因组新技术,目前已经广泛应用在全基因组关联分析(GWAS)、遗传图谱构建及QTL定位、群体遗传进化分析和基因组选择等领域[18-21]。本研究同时采用线粒体DNA控制区(D-loop)片段和SLAF-seq技术对中国沿海多室草苔虫的群体遗传分化开展研究,研究结果对深入了解这种重要药源生物在我国的资源状况及种群分化具有重要意义。
2 材料与方法
2.1 样品采集与DNA提取
根据中国多室草苔虫的分布范围及可能存在的地理阻隔特征,本研究采集了4个地理群体共104个个体(表1),分别来自于三亚(SY)、深圳(SZ)、厦门(XM)和温州(WZ)(图1)。所有个体都根据《中国海洋污损苔虫生物学》一书中的分类检索表进行鉴定。样品用无菌海水清洗3次后,置于95%的乙醇中固定,-20℃保存。
DNA的提取参考孙名安等[22]关于苔藓动物基因组DNA的提取方法。提取的DNA经琼脂糖凝胶电泳检测合格后于-20℃保存备用。
2.2线粒体DNA控制区片段克隆和遗传多样性分析
根据GenBank上的多室草苔虫线粒体全基因组序列(序列号:NC_010197),利用Primer Premier 5软件设计扩增D-loop片段的引物。引物F-bncn序列为5′-CTAATTTCTCACCCTTATTC-3′,引物R-bncn序列为5′-GTTGGTTGGCTGACACTT-3′。

图1 多室草苔虫采样地点示意图Fig.1 Sampling localities of B. neritina
PCR的反应体系为25 μL,其中包括10×反应缓冲液(Mg2+)2.5 μL, 引物 (10 μmol/L)各0.5 μL,Taq酶(2.5 U/μL) 0.2 μL, DNA 模板(100 ng/μL)1 μL, dNTPs(2.5 mmol/L)2 μL, ddH2O 18.3 μL。PCR反应条件为:94℃预变性5 min;94℃变性30 s,53℃退火30 s,72℃延伸90 s,共30个循环;72℃延伸10 min。PCR产物使用TaKaRa DNA Fragment Purification kit纯化后进行凝胶电泳检测,扩增结果良好的样品送至北京六合华大基因科技股份公司进行测序。
测序结果使用Sequencher 4.1.4软件进行比对和校正,使用DnaSP 5.0软件[23]统计单倍型数目,并计算单倍型多样性和核苷酸多样性。使用Network 5.0.0.0软件构建单倍型网络图。采用 Tajima′sD和Fu′sFs中性检验以及核苷酸不配对分布来检测多室草苔虫群体的历史动态。
2.3 SLAF-seq简化基因组测序
2.3.1 酶切建库
选取紫色球海胆(Strongylocentrotuspurpuratus)基因组作为参考基因组进行酶切预测。根据最优酶切方案,从三亚(SY)、深圳(SZ)、厦门(XM)和温州(WZ)群体中各随机选取12个样品对其基因组DNA进行酶切,对得到的SLAF标签3′端进行加A处理,连接Dual-index测序接头后PCR扩增,纯化,产物混样处理后切胶回收目的大小片段的产物。
2.3.2 高通量测序及测序片段质量检验和分析
文库构建成功质检合格后用Illumina HiSeq 2500进行测序。选取拟南芥(Arabidopsisthalianaecotype Columbia)参与酶切和测序作为对照。利用Dual-index对测序得到的原始数据进行识别,得到各个样品的reads。过滤掉测序reads的接头后,对其进行GC含量和Q30分析,以评估测序质量和数据量。通过对照的数据评估选取的酶切方案的酶切效率,判断实验的准确性和有效性。
2.3.3SLAF标签的获得和全基因组范围SNP标记的开发
根据序列相似度对各个样品的reads进行聚类,聚类到一起的reads来源于同一个SLAF标签,同一个SLAF标签在不同样品间的序列相似度要远高于不同SLAF标签间的相似度。一个SLAF标签在不同样品间有差异即可定义为多态性SLAF标签。根据具有多态性的SLAF标签寻找SNP位点。
2.4 群体遗传结构分析
基于线粒体DNA控制区片段的测序结果,使用MEGA 5.0软件[24]计算群体内和群体间基于Kimura 2-parameter的遗传距离,并采用邻接法构建单倍型的系统进化树,设定重复抽样1 000次以检验单倍型关系树各分枝的置信度。使用Arlequin 3.5.1.2软件[25]中的分子方差分析(AMOVA)估算遗传变异的分布和固定系数Fst,1 000次随机重复抽样后进行显著性检验。
从SLAF-seq技术开发得到的分子标记中,选取群体内具有代表性的高质量(筛选标准为MAF>0.05)的SNP位点开展群体遗传结构分析。使用MEGA 5软件[24],运用neighbor-joining算法[26],构建群体的进化树。使用structure软件[27]和admixture软件[28],分别假设样品的分群数(K值)为1~10,进行聚类,通过交叉验证确定分群数为何时验证结果的错误率ΔK最低,以ΔK值最低的分群作为最佳分群数。使用cluster软件[29],进行主元成分分析(Principal components analysis, PCA)分析,通过样品间的接近程度辅助进化分析。
3 结果
3.1 基于线粒体控制区序列的结果
3.1.1 遗传多样性分析和单倍型统计
共检测到10个多态简约信息位点和11个核苷酸变异位点,定义了8个单倍型。单倍型多样性(h)和核苷酸多样性(π)分别为0.130 7和0.000 7,显示其多样性水平较低。各个群体的遗传多样性参数统计情况见表1。
单倍型网络图如图2所示。Hap 1是所有群体共享的主单倍型。Hap 5、Hap 6、Hap 7这3个单倍型均是三亚群体的独有单倍型,且数量仅有1个。
4个群体D-loop片段的单倍型NJ系统树,如图3所示。从图中可以看出进化枝主要分为两支,但两进化枝之间枝长均较短。
3.1.2 群体历史动态
各群体和总体的Fu′sFs和Tajima′sD中性检验结果如表2所示。结果显示,除了三亚群体的Fu′sFs中性结果为正值外,其他的都为负值,然而除了深圳群体的结果为显著以外,其他群体和总体的中性检验结果都不显著。4个群体的核苷酸不配对分析和吻合度分析见表2,根据核苷酸不配对分析结果所做的核苷酸不配对分布曲线如图4所示。结果显示,4个群体的核苷酸不配对分布曲线都不是典型的单峰分布,说明多室草苔虫在历史上可能并未发生过大规模扩张事件。

表1 不同群体的遗传多样性参数统计
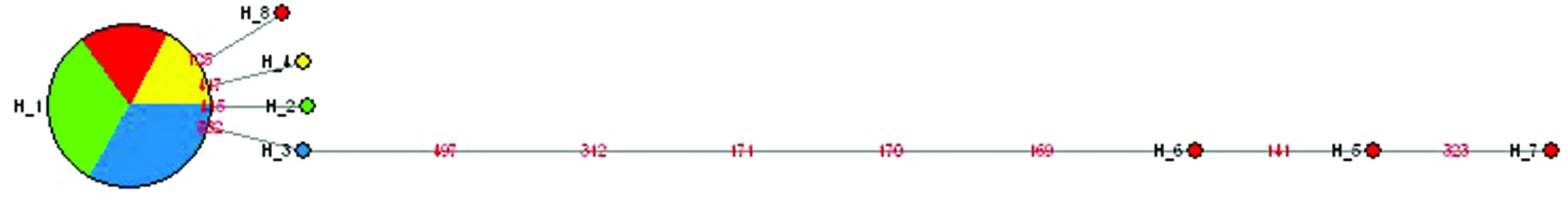
图2 多室草苔虫4个群体的单倍型网络图Fig.2 Haplotype network of 4 B. neritina populations
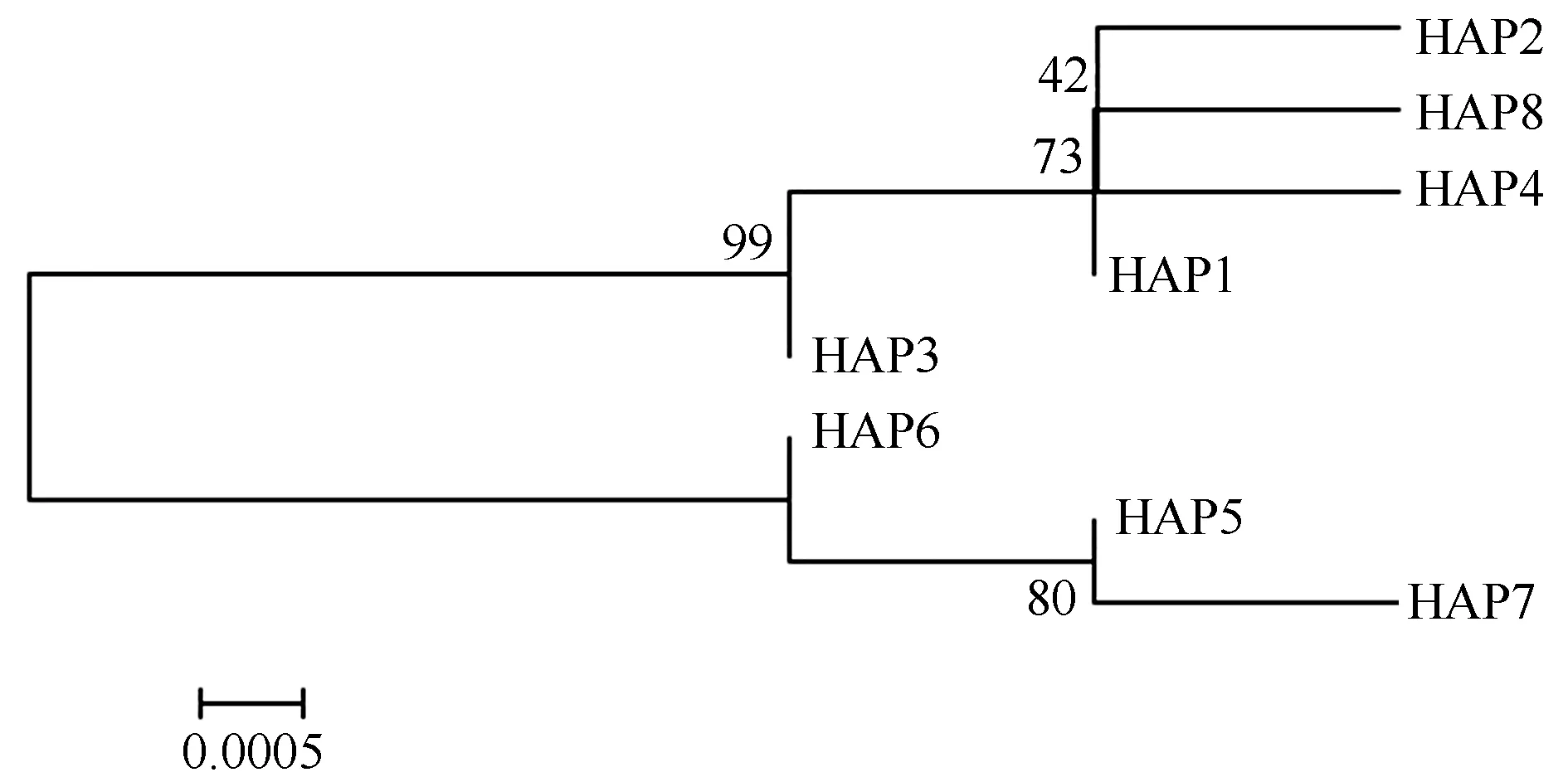
图3 多室草苔虫4个群体的D-loop片段的单倍型NJ系统树Fig.3 Neighbor-joining tree of D-loop sequence haplotypes from 4 B. neritina populations
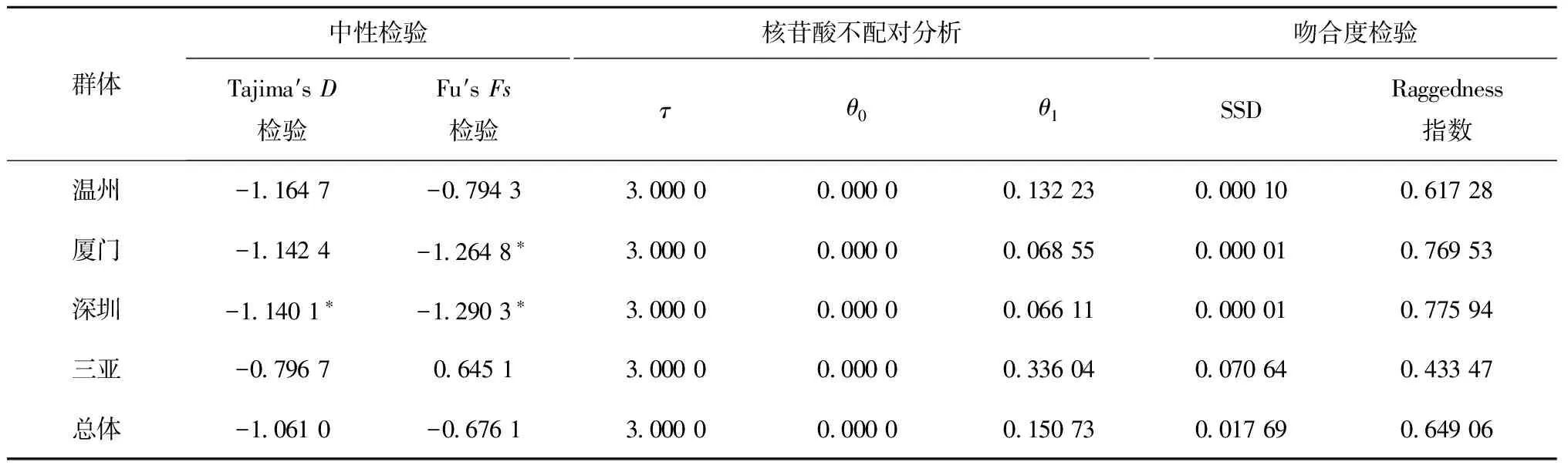
群体中性检验核苷酸不配对分析吻合度检验Tajima′sD检验Fu′sFs检验τθ0θ1SSDRaggedness指数温州-11647-079433000000000013223000010061728厦门-11424-12648∗3000000000006855000001076953深圳-11401∗-12903∗3000000000006611000001077594三亚-07967064513000000000033604007064043347总体-10610-067613000000000015073001769064906
注:τ 用突变单位表示的种群扩张时间,θ0种群开始扩张前大小,θ1种群扩张后大小;数值右上方的*号表示结果显著。
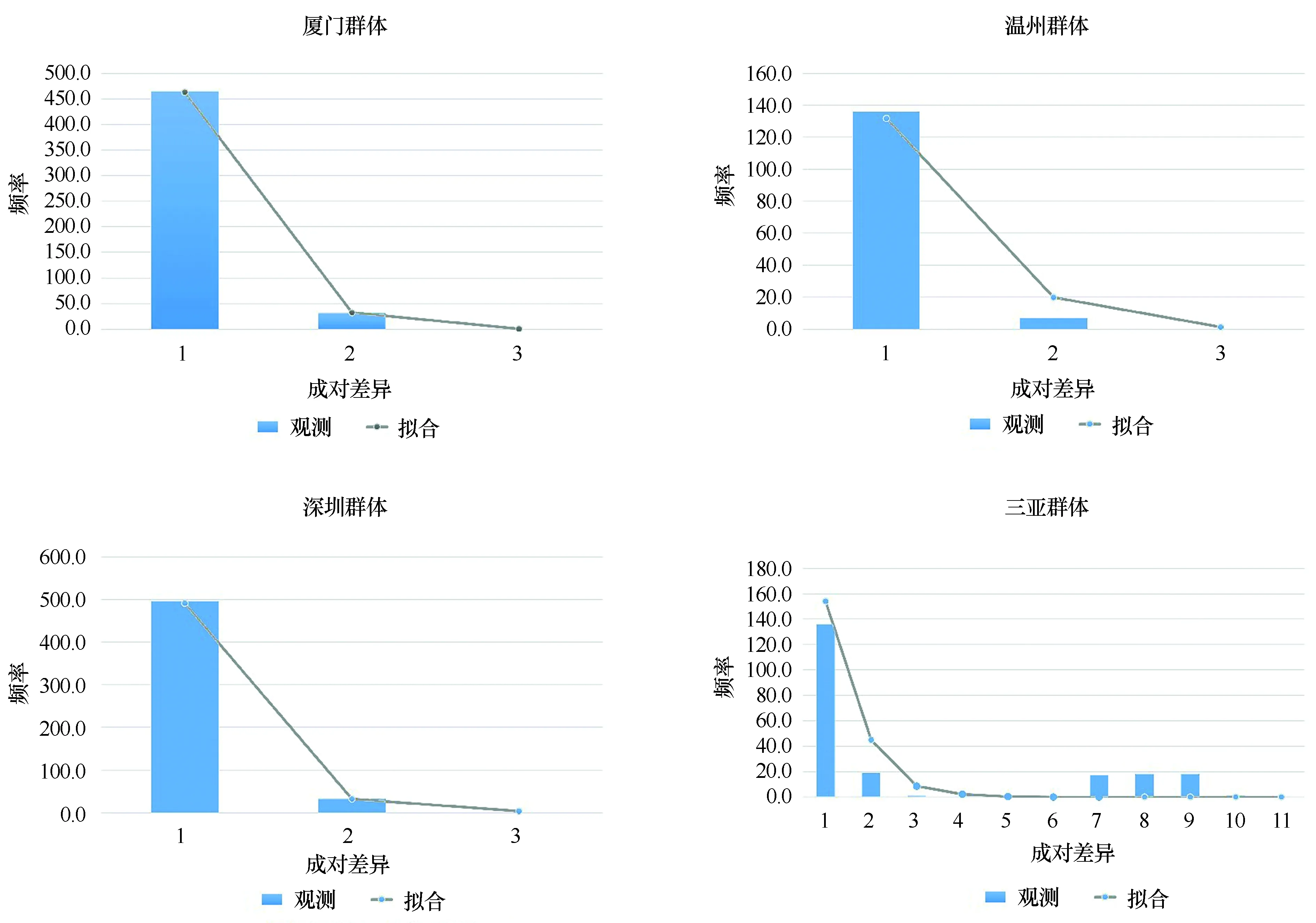
图4 多室草苔虫4个群体D-loop序列核苷酸不配对分布图Fig.4 Mismatch distribution for D-loop sequences of 4 B. neritina populations
3.2 基于SLAF-seq简化基因组的结果
3.2.1 SLAF建库评估
根据对参考基因组序列的电子酶切结果,确定最佳的限制性内切酶组合为HinCII+EcoRV-HF®,酶切长度在314~344 bp的序列定义为SLAF标签,预测可得到14 540个SLAF标签。另外,拟南芥的酶切片段双端比对效率为79.21%,表明比对效率基本正常;酶切效率为99.21%,表明酶切反应正常。综上所述,本实验SLAF建库正常。
3.2.2 测序数据统计与评估
为保证后续生物信息学分析质量,实验采用去掉接头后读长的80 bp×2作为后续的数据评估和分析数据。各样品经测序共获得11.93 M的reads数据,每个样品的总测序读长74 430~735 578 bp不等。各样品的GC含量35.09%~48.85%,达到测序要求;来自所有样品的个体碱基测序评估的指标Q30比例80.01%~91.16%,所有样品Q30比例都在80%以上,说明测序过程中碱基错误率低,获得的测序数据质量合格可信。
3.2.3 SLAF标签的开发和SNP信息统计
实验中针对所有样品共开发得到214 409个SLAF标签,样品的平均测序深度为10.29×。根据等位基因数和基因序列之间的差异进行多态性分析,共得到23 437个多态性SLAF标签,比例为10.93%;190 972个非多态性的SLAF标签,比例为89.07%。利用得到的多态性SLAF标签共开发出99 432个SNP位点。
3.3 多室草苔虫群体遗传结构分析
基于控制区序列的4个群体的遗传距离,如表3所示。从中可以看出,群体内遗传距离的大小比较为三亚(0.002 82)>温州(0.000 16)>深圳(0.000 09)=厦门(0.000 09),而群体间的遗传距离范围为0.000 09~0.001 62。整体来看,群体间的遗传距离水平低于群体内的遗传距离,没有显著的遗传结构。但是,三亚群体与另外3个群体之间的遗传距离相对较远。
注:*为群体内Kimura 2-parameter遗传距离。
基于控制区序列的4个群体的遗传分化,其结果如表4所示。Wright[30]曾经提出了关于遗传分化指数的大小和分化程度的解释,他认为Fst值小于0.05时,群体间遗传分化程度很低;在0.05~0.15之间表明群体遗传分化达中等水平。由基于D-loop片段序列的数据可知,三亚群体对另外3个群体的Fst值均高于0.05,且三亚对深圳和三亚对厦门的Fst分析均显示差异显著。这说明三亚群体相对于另外3个群体可能存在一定的遗传分化。

表4 多室草苔虫4个群体的遗传分化
注:对角线以下群体间Fst值;对角线以上基于Fst值的显著性分析(以P<0.05为标准)结果差异显著为“+”,反之为“-”。
将三亚群体和另外3个群体分别归为1个群组进行AMOVA分析,结果如表5所示。可以看出,有22.75%的变异来自群组间,而79.70%的变异来自群体内,而群体间、群组间固定系数Fst和Fct分别为0.095 12和0.227 47,说明三亚群体和其他群体间存在较大的遗传差异。

表5 多室草苔虫群组间的AMOVA分析
基于SNP位点的系统发育树见图5。由图可知,系统发育树呈现出与分布地点不相关的散状分枝,说明各群体之间的遗传分化并不明显。
基于SNP位点的群体遗传结构的分析结果显示,来自不同地点的群体的最佳分群数为1(图6,图7),这表明所有多室草苔虫均属于同一个群体。PCA分析结果(图8)则显示,来自温州、厦门、深圳的样品比较接近,而来自三亚的样品与其他地点的样品较为疏远。
4 讨论
4.1多室草苔虫的遗传多样性水平和群体遗传结构分析
物种的遗传多样性水平与其适应和生存的能力以及进化的潜力密切相关[31]。基于线粒体控制区序列的结果表明,我国的多室草苔虫群体的单倍型多样性(h)和核苷酸多样性(π)均处于较低水平。当种群的h值和π值都较低时(h<0.5,π<0.005),说明该种群的遗传多样性水平较低,有可能在进化过程中经历了瓶颈效应(bottleneck effect)或者是奠基者效应(founder effect)[32]。群体历史动态结果也显示多室草苔虫在历史上没有发生过显著的扩张行为。基于SLAF-seq的群体遗传分化分析结果显示,只有约10%的SLAF标签是多态性的,这同样表明该物种的遗传多样性水平较低。由此,基于线粒体DNA和SLAF-seq的群体遗传分化结果都一致表明,多室草苔虫的遗传多样性处于较低的水平,这也与其他学者对世界范围内多室草苔虫的群体遗传学研究中只发现了1种COⅠ单倍型的结果相吻合[33]。

图5 基于SNP位点的系统进化树Fig.5 Phylogenetic tree based on SNP loci
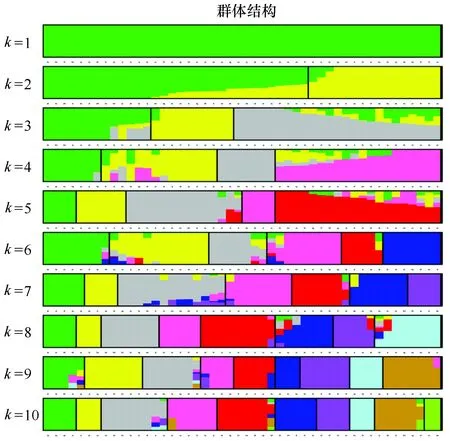
图6 分群数为1~10的聚类图Fig.6 Dendrogram of clustering from 1-10
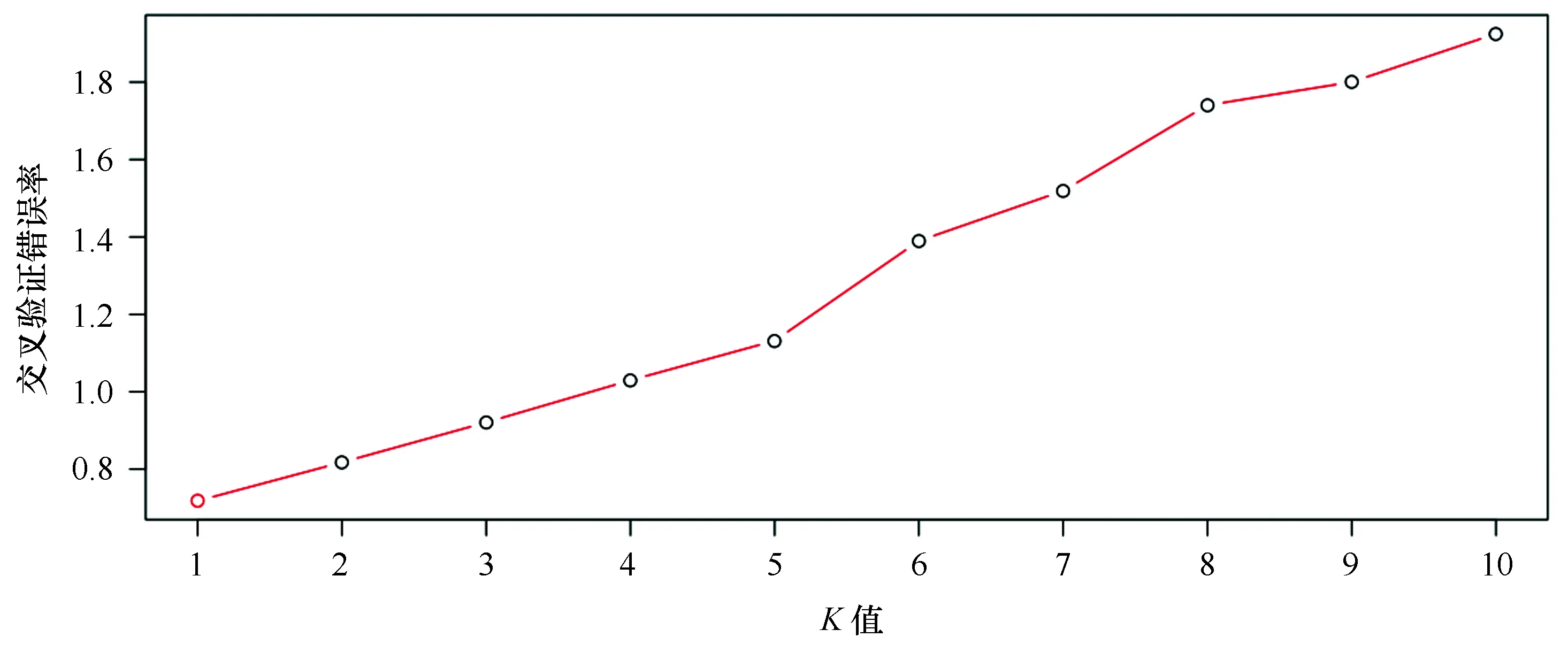
图7 ΔK值分布图Fig.7 Scattergram of ΔK value

图8 PCA聚类分析图Fig.8 PCA clustering analysis
群体遗传结构分析结果表明,4个不同地点的多室草苔虫之间未检测到显著的遗传结构,故应属于同一个群体。导致生物群体遗传结构同质化的原因有很多,对于海洋生物而言,其自身的生活史与扩散能力、海洋水文环境条件和理化因素、生境的连续性等,都是有可能影响其遗传结构的重要因素[34]。
多室草苔虫是固生的海洋动物,其完整生活史中只有幼虫阶段具有潜在的扩散能力,但幼虫在海水中仅能自由游动1~2 h便会附着在固着基上[35-36],其较差的扩散能力,限制了不同地点群体间的基因交流[37-38]。在凭借自身条件难以实现远距离扩散的情况下,多室草苔虫仍可以依靠舰船航行扩散。舰船航行使得依附在其表面的污损生物有可能克服其原本无法逾越的海洋陆地阻隔、温度差别以及低盐度的河口径流等障碍,从而扩大种群的分布范围[1]。然而,要实现上述目的污损生物还必须满足以下条件:(1)能经受舰船快速航行和波浪冲击;(2)能抵御温度、盐度等环境条件的急剧变化;(3)能在新环境里进行有性生殖[39]。对多室草苔虫而言,其直立生长和钙化程度较低的群体特性不利于应对舰船快速航行和波浪冲击,但其幼虫附着变态后幼体生长发育迅速,形成初虫直立生长至群体第四次分歧仅需30 d,因此,多室草苔虫仍然可借助附着在舰船底部进行有效的长距离扩散[1]。另外一方面,作为一种常见的污损生物,多室草苔虫对栖息环境的适应范围较广,在半裸露的岩石、浮标、栈桥、渔排、船底、海带和石花菜表面等地方均可大量生长[1],在扩散到新的环境后有能力生存下来。
综上所述,多室草苔虫广泛的扩散行为和对环境较好的适应性可以使不同群体间保持频繁的基因交流,在一定程度上使基因库趋于同质化,同时抵消了选择压力下对特定遗传变异的保留[40],进而影响群体的遗传多样性水平并阻碍群体遗传结构的产生。
4.2SLAF-seq应用在多室草苔虫群体遗传分化研究中的可行性和优势
目前,SLAF-seq技术已经在各个领域的研究中成功开发出众多分子标记[41]。对于没有参考基因组的物种,如鲤鱼等,SLAF-seq也能进行SNP位点的挖掘[42];对于基因组序列组成高度杂合的物种,如牡丹、茶树、胡桃等,在没有参考基因组序列的情况下,SLAF-seq可以通过先进行电子酶切设计方案再测序的方式,实现分子标记的大规模开发[43-45];而对于多态率低的物种,如大豆等,SLAF-seq大规模开发分子标记的能力也得到过检验[18]。因此,尽管目前由于没有多室草苔虫及其近缘物种的基因组序列可供参考,无法了解其基因组的复杂程度和组成特性,但是根据SLAF-seq技术在其他诸多物种上的成功实践案例,我们依然可以将其用于多室草苔虫全基因组范围内SNP位点的开发。
分子标记开发的结果显示,实际开发出的SLAF标签数远高于用电子酶切预测的结果,同时做到了对所有样品的深度覆盖。10.29×的覆盖率,不仅高于预期的5×,同时也确保了碱基读取的准确性和SNP位点的有效性。另外,获得的SLAF标签中没有重复性标签,而只有多态性标签和非多态性标签。由于缺乏参考基因组信息,无法排除多室草苔虫基因组本身重复序列比例较低的可能性;但是事先通过电子酶切来预测最优酶切方案,显然对于研究结果中重复性标签的杜绝和分子标记挖掘效率的提升起到了重要的作用。最终开发出的99 432个分子标记,数量可观,并实现了在全基因组范围内的高密度均匀覆盖,因此完全可以用于后续的群体遗传分析。另外, SLAF-seq在遗传多样性水平和群体遗传结构方面,呈现出与线粒体DNA序列相一致的结果,这也从另一个侧面证明了,SLAF-seq技术完全可以应用于包括多室草苔虫在内的海洋生物群体遗传分化研究。
作为一种生物信息学分析辅助的基于酶切的简化基因组技术,SLAF-seq有着(1)不受参考基因组限制,可以应用于各种非模式生物;(2)对SLAF标签的深度测序可以保证基因分型的高度准确性;(3)对目标基因组简化代表的策略显著降低了测序成本;(4)酶切方案灵活,能够有效避开重复序列,优化了分子标记的挖掘效率;(5)开发的SNP分子标记性价比高,稳定性好,在基因组中分布均匀;(6)双端barcode接头的系统非常适合大群体研究等诸多优势[42,46]。特别是相较于其他传统的分子标记,如线粒体DNA等,SLAF-seq有着高出几个数量级的分子标记数目。另外,由于在全基因组范围内的高密度均匀覆盖,而非其它分子标记仅仅来源于基因组的局部有限范围,如线粒体基因组等,SLAF-seq在群体研究中有着更精细的分辨率,因而能传递出更多的遗传信息。在本研究中,尽管两种技术手段的结果基本一致,但是相较于线粒体控制区序列结果中的8个单倍型,SLAF-seq从来源相同、样品数目却更少的4个群体中共得到99 432个SNP位点,其呈现出的有关遗传变异的信息量显然更为丰富和准确,因此,能够更加全面和深入地反映多室草苔虫的群体遗传结构和群体遗传分化水平。另外,SLAF-seq在群体遗传学研究中对每个群体的样品数量要求较低,在综合考虑研究结果的准确性和测序成本的前提下,通常只需要10~15个,较线粒体DNA等传统分子标记所需的30~50个而言大幅减少,在一定程度上降低了采样成本和难度。由此可见,相较于线粒体DNA等传统分子标记,SLAF-seq技术在群体遗传分化研究中有着无可比拟的巨大技术优势。
[1] 刘锡兴, 尹学明, 马江虎. 中国海洋污损苔虫生物学[M]. 北京: 科学出版社, 2001: 79-467.
Liu Xixing, Yin Xueming, Ma Jianghu. Biology of Marine-fouling Bryozoans in the Coastal Waters of China[M]. Beijing: Science Press, 2001: 79-467.
[2] 林厚文, 易杨华, 姚新生, 等. 中国南海总合草苔虫抗癌活性成分研究(Ⅳ) Bryostatin 8, 16的分离与结构鉴定[J]. 中国海洋药物, 2001, 20(4): 1-6.
Lin Houwen, Yi Yanghua, Yao Xinsheng, et al. Studies on antineoplastic constituents from marine bryozoanBugulaneritinainhabiting South China Sea (Ⅳ): isolation and structural elucidation of bryostatins 8 and 16[J]. Chinese Journal of Marine Drugs, 2001, 20(4): 1-6.
[3] 林厚文, 易杨华, 姚新生, 等. 中国南海总合草苔虫抗癌活性成分研究——Ⅱ总草苔虫内酯的超强抗癌活性[J]. 中国海洋药物, 2000, 19(2): 1-3.
Lin Houwen, Yi Yanghua, Yao Xinsheng, et al. Studies on the antineoplastic constituents from marine bryozoanBugulaneritinainhabiting South China Sea (Ⅱ): remarkable antineoplastic activities of active principals[J]. Chinese Journal of Marine Drugs, 2000, 19(2): 1-3.
[4] Lei Hui, Zhou Xuefeng, Yang Yaling, et al. Bryostatins from South China Sea bryozoanBugulaneritinaL.[J]. Biochemical Systematics and Ecology, 2010, 38(6): 1231-1233.
[5] Lopanik N, Gustafson K R, Lindquist N. Structure of bryostatin 20: a symbiont-produced chemical defense for larvae of the host bryozoan,Bugulaneritina[J]. Journal of Natural Products, 2004, 67(8): 1412-1414.
[6] 孙鹏, 李玲, 易杨华, 等. 总合草苔虫中抗癌活性成分的提取和含量测定[J]. 第二军医大学学报, 2002, 23(3): 240-242.
Sun Peng, Li Ling, Yi Yanghua, et al. Extraction and quantitative determination of antineoplastic constituents inBugulaneritinaL.[J]. Academic Journal of Second Military Medical University, 2002, 23(3): 240-242.
[7] Kraft A S, Smith J B, Berkow R L. Bryostatin, an activator of the calcium phospholipid-dependent protein kinase, blocks phorbol ester-induced differentiation of human promyelocytic leukemia cells HL-60[J]. Proceedings of the National Academy of Sciencesof the United States of America, 1986, 83(5): 1334-1338.
[8] Kuzirian A M, Epstein H, Gagliardi C J, et al. Bryostatin enhancement of memory inHermissenda[J]. The Biological Bulletin, 2006, 210(3): 201-214.
[9] Parkinson D R, Arbuck S G, Moore T, et al. Clinical development of anticancer agents from natural products[J]. Stem Cells, 1994, 12(1): 30-43.
[10] Sun Miaokun, Alkon D L. Bryostatin-1: pharmacology and therapeutic potential as a CNS drug[J]. CNS Drug Reviews, 2006, 12(1): 1-8.
[11] Sun Miaokun, Alkon D L. Dual effects of bryostatin-1 on spatial memory and depression[J]. European Journal of Pharmacology, 2005, 512(1): 43-51.
[12] 林厚文, 易杨华, 李文林, 等. 中国南海总合草苔虫中新的抗癌活性成分Bryostatin19[J]. 中国海洋药物, 1998, 17(1): 1-3.
Lin Houwen, Yi Yanghua, Li Wenlin, et al. Bryostatin 19: a new antineoplastic component fromBugulaneritinain the South China Sea[J]. Chinese Journal of Marine Drugs, 1998, 17(1): 1-3.
[13] Pettit G R, Kamano Y, Herald C L, et al. Isolation and structure of bryostatins 5-7[J]. Canadian Journal of Chemistry, 1985, 63(6): 1204-1208.
[14] 曹艳, 章群, 宫亚运, 等. 基于线粒体COⅠ序列的中国沿海蓝点马鲛遗传多样性[J]. 海洋渔业, 2015, 37(6): 485-493.
Cao Yan, Zhang Qun, Gong Yayun, et al. Genetic variation ofScomberomorusniphoniusin the coastal waters of China based on mt DNA COⅠ sequences[J]. Marine Fisheries, 2015, 37(6): 485-493.
[15] 刘若愚, 孙忠民, 姚建亭, 等. 中国近海重要生态建群红藻真江蓠的群体遗传多样性[J]. 生物多样性, 2016, 24(7): 781-790.
Liu Ruoyu, Sun Zhongmin, Yao Jianting, et al. Genetic diversity of the habitat-forming red algaGracilariavermiculophyllaalong Chinese coasts[J]. Biodiversity Science, 2016, 24(7): 781-790.
[16] Guo Xiang, Zhao Dan, Jung D, et al. Phylogeography of the rock shellThaisclavigera(Mollusca): evidence for long-distance dispersal in the northwestern Pacific[J]. PLoS One, 2015, 10(7): e0129715.
[17] Wang Jie, Tsang L M, Dong Yunwei. Causations of phylogeographic barrier of some rocky shore species along the Chinese coastline[J]. BMC Evolutionary Biology, 2015, 15: 114.
[18] Li Bin, Tian Ling, Zhang Jingying, et al. Construction of a high-density genetic map based on large-scale markers developed by specific length amplified fragment sequencing (SLAF-seq) and its application to QTL analysis for isoflavone content inGlycinemax[J]. BMC Genomics, 2014, 15: 1096.
[19] Wang Wenhao, Zhang Tao, Zhang Genxi, et al. Genome-wide association study of antibody level response to NDV and IBV in Jinghai yellow chicken based on SLAF-seq technology[J]. Journal of Applied Genetics, 2015, 56(3): 365-373.
[20] 陈士强, 秦树文, 黄泽峰, 等. 基于SLAF-seq技术开发长穗偃麦草染色体特异分子标记[J]. 作物学报, 2013, 39(4): 727-734.
Chen Shiqiang, Qin Shuwen, Huang Zefeng, et al. Development of specific molecular markers forThinopyrumelongatumchromosome using SLAF-seq technique[J]. Acta Agronomica Sinica, 2013, 39(4): 727-734.
[21] 苏文瑾, 赵宁, 雷剑, 等. 基于SLAF-seq技术的甘薯SNP位点开发[J]. 中国农业科学, 2016, 49(1): 27-34.
Su Wenjin, Zhao Ning, Lei Jian, et al. SNP sites developed by specific length amplification fragment sequencing (SLAF-seq) in Sweetpotato[J]. Scientia Agricultura Sinica, 2016, 49(1): 27-34.
[22] 孙名安, 吴志刚, 申欣, 等. 颈链血苔虫线粒体基因组的测定及其系统发育学意义[J]. 渔业科学进展, 2010, 31(1): 89-94.
Sun Ming’an, Wu Zhigang, Shen Xin, et al. The complete mitochondrial genome ofWatersiporasubtorquataand its phylogenetic significance[J]. Progress in Fishery Sciences, 2010, 31(1): 89-94.
[23] Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data[J]. Bioinformatics, 2009, 25(11): 1451-1452.
[24] Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods[J]. Molecular Biology and Evolution, 2011, 28(10): 2731-2739.
[25] Excoffier L, Lischer H E L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows[J]. Molecular Ecology Resources, 2010, 10(3): 564-567.
[26] Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees[J]. Molecular Biology and Evolution, 1987, 4(4): 406-425.
[27] Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study[J]. Molecular Ecology, 2005, 14(8): 2611-2620.
[28] Alexander D H, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals[J]. Genome Research, 2009, 19(9): 1655-1664.
[29] de Hoon M J L, Imoto S, Nolan J, et al. Open source clustering software[J]. Bioinformatics, 2004, 20(9): 1453-1454.
[30] Wright S. The genetical structure of populations[J]. Annals of Eugenics, 1949, 15(1): 323-354.
[31] 牛素芳, 苏永全, 王军, 等. 福建近海蓝圆鲹群体遗传结构分析[J]. 厦门大学学报:自然科学版, 2012, 51(4): 759-766.
Niu Sufang, Su Yongquan, Wang Jun, et al. Population genetic structure analysis ofDecapterusmaruadsifrom Fujian coastal waters[J]. Journal of Xiamen University:Natural Science, 2012, 51(4): 759-766.
[32] Grant W A S, Bowen B W. Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation[J]. Journal of Heredity, 1998, 89(5): 415-426.
[33] Mackie J A, Keough M J, Christidis L. Invasion patterns inferred from cytochrome oxidase Ⅰ sequences in three bryozoans,Bugulaneritina,Watersiporasubtorquata, andWatersiporaarcuata[J]. Marine Biology, 2006, 149(2): 285-295.
[34] 张丽艳. 台湾海峡三种中上层鱼类遗传多样性的AFLP分析[D]. 厦门: 厦门大学, 2011.
Zhang Liyan. Genetic diversity of three pelagic fishes in the Taiwan Strait, inferred by AFLP fingerprinting[D]. Xiamen: Xiamen University, 2011.
[35] Dahms H U, Dobretsov S, Qian Peiyuan. The effect of bacterial and diatom biofilms on the settlement of the bryozoanBugulaneritina[J]. Journal of Experimental Marine Biology and Ecology, 2004, 313(1): 191-209.
[36] Wendt D E. Energetics of larval swimming and metamorphosis in four species ofBugula(Bryozoa)[J]. The Biological Bulletin, 2000, 198(3): 346-356.
[37] Keough M J. Dispersal of the bryozoanBugulaneritinaand effects of adults on newly metamorphosed juveniles[J]. Marine Ecology Progress Series, 1989, 57: 163-171.
[38] Wendt D E. Effect of larval swimming duration on success of metamorphosis and size of the ancestrular lophophore inBugulaneritina(Bryozoa)[J]. The Biological Bulletin, 1996, 191(2): 224-233.
[39] 黄宗国, 蔡如星. 海洋污损生物及其防除[M]. 北京: 海洋出版社, 1984: 1-352.
Huang Zongguo, Cai Ruxing. Marine Fouling and Its prevention[M]. Beijing: China Ocean Press, 1984: 1-352.
[40] Guo Baocheng, de Faveri J, Sotelo G, et al. Population genomic evidence for adaptive differentiation in Baltic Sea three-spined sticklebacks[J]. BMC Biology, 2015, 13: 19.
[41] Wei Qingzhen, Wang Yunzhu, Qin Xiaodong, et al. An SNP-based saturated genetic map and QTL analysis of fruit-related traits in cucumber using specific-length amplified fragment (SLAF) sequencing[J]. BMC Genomics, 2014, 15: 1158.
[42] Sun Xiaowen, Liu Dongyuan, Zhang Xiaofeng, et al. SLAF-seq: an efficient method of large-scaledenovoSNP discovery and genotyping using high-throughput sequencing[J]. PLoS One, 2013, 8(3): e58700.
[43] Cai Changfu, Cheng Fangyun, Wu Jing, et al. The first high-density genetic map construction in tree peony (PaeoniaSect.Moutan) using genotyping by specific-locus amplified fragment sequencing[J]. PLoS One, 2015, 10(5): e0128584.
[44] Ma Jianqiang, Huang Long, Ma Chunlei, et al. Large-scale SNP discovery and genotyping for constructing a high-density genetic map of tea plant using specific-locus amplified fragment sequencing (SLAF-seq)[J]. PLoS One, 2015, 10(6): e0128798.
[45] Zhu Yufeng, Yin Yanfei, Yang Keqiang, et al. Construction of a high-density genetic map using specific length amplified fragment markers and identification of a quantitative trait locus for anthracnose resistance in walnut (JuglansregiaL.)[J]. BMC Genomics, 2015, 16: 614.
[46] Zheng Wenjing, Li Zhiqiang, Zhao Jiaming, et al. Study of the long-distance migration of small brown planthoppersLaodelphaxstriatellusin China using next-generation sequencing[J]. Pest Management Science, 2016, 72(2): 298-305.
Population genetic variation study of Bugula neritina in coastal waters of China
Li Hai1,2,Liu Qiaohong2,Tang Xueying2,Chen Wuge3,Ding Shaoxiong2
(1.LaboratoryofMarineBiologyandEcology,ThirdInstituteofOceanography,StateOceanicAdministration,Xiamen361005,China; 2.FujianCollaborativeInnovationCenterforExploitationandUtilizationofMarineBiologicalResources,Xiamen361102,China; 3.XiamenOceanVocationalCollege,Xiamen361012,China)
We studied the genetic diversity and population genetic variation ofBugulaneritina,an important pharmaceutical organism,sampled from 4 distinct localities along China coastline by mitochondrial control region amplification and SLAF-seq. 8 haplotypes were detected in control region, haplotype diversity (h) and nucleotide diversity (π) were 0.130 7 and 0.000 7, respectively. No significant topological structure was found in haplotype network and NJ phylogenetic tree. Neutrality tests and mismatch distribution both suggested thatB.neritinadid not experience a range expansion.Fstand AMOVA analysis indicated that genetic variation mainly occurred within populations. SLAF library construction generated 214 409 SLAFs, among which 23 437 were polymorphic, 99 432 SNP loci were developed. Genetic distances among populations were short and even shorter than those within populations. Phylogenetic tree and population genetic analysis based on SNP data revealed that no significant genetic structure were observed among populations. In conclusion, genetic diversity ofB.neritinain coastal waters of China were low, and no significant genetic structure existed among geographically distinct populations. We assumed this was mainly attributed to bryozoan’s capability to disperse. In addition, our study validated the application and advantages of SLAF-seq in population genetic variation study of marine organisms.
Bugulaneritina; population genetic variation; mitochondrial DNA; SLAF-seq; reduced representation sequencing
10.3969/j.issn.0253-4193.2017.10.008
Q953+.1
:A
:0253-4193(2017)10-0090-11
2017-02-28;
:2017-06-15。
国家海洋局公益性项目“几种重要海洋药用生物种质资源发掘、保藏和利用——重要海洋药用生物优良种质评价和筛选”(201205024-2)。
李海(1988—),男,江西省萍乡市人,博士,主要从事海洋生物遗传多样性与进化研究。E-mail:lihai@tio.org.cn
*通信作者:丁少雄,教授,主要从事海洋生物资源保护与利用研究。E-mail:sxding@xmu.edu.cn
李海,刘巧红,唐雪颖,等. 我国近岸多室草苔虫(Bugulaneritina)的群体遗传分化研究 [J].海洋学报,2017,39(10):90—100,
Li Hai,Liu Qiaohong,Tang Xueying,et al. Population genetic variation study ofBugulaneritinain coastal waters of China[J]. Haiyang Xuebao,2017,39(10):90—100, doi:10.3969/j.issn.0253-4193.2017.10.008
