珠子参药材中皂苷类成分的薄层色谱及HPLC特征图谱研究△
2017-09-22张海元夏伟军谢佳颖唐梦云张英杰崔蓉梅双喜杨立国
张海元,夏伟军,谢佳颖,唐梦云,张英杰,崔蓉,梅双喜,杨立国
(云南省药物研究所 云南白药集团创新研发中心 云南省中药和民族药新药创制企业重点实验室,云南 昆明 650111)
·中药基础·
珠子参药材中皂苷类成分的薄层色谱及HPLC特征图谱研究△
张海元,夏伟军,谢佳颖,唐梦云,张英杰,崔蓉,梅双喜,杨立国*
(云南省药物研究所 云南白药集团创新研发中心 云南省中药和民族药新药创制企业重点实验室,云南 昆明 650111)
目的:建立和完善珠子参药材的薄层色谱及HPLC特征图谱鉴别方法。方法:采用薄层色谱法鉴别珠子参药材中皂苷类成分;采用HPLC法对同一产地不同地块的10批次珠子参药材皂苷类成分进行指纹图谱分析,选用Waters XTerra MS C18色谱柱(250 mm×4.6 mm,5 μm);流动相为乙腈-0.2%磷酸水溶液,梯度洗脱,分析时间为55 min,检测波长为203 nm。结果:珠子参药材的薄层色谱中皂苷类成分分离度好,斑点清晰,专属性强,同时建立了珠子参皂苷类成分的HPLC特征图谱方法。结论:薄层色谱及HPLC特征图谱方法稳定、可靠、重复性好,为珠子参药材的质量控制提供了科学依据。
珠子参,皂苷成分,薄层色谱,HPLC特征图谱
五加科植物珠子参PanaxjaponicusC.A.Mey.var.major(Burk.)C.Y.Wu et K.M.Feng主产于四川、云南、贵州、陕西、甘肃、西藏等地,其干燥根茎入药,又名扣子七、纽子七、珠儿参。珠子参具有补肺、养阴、活络、止血功效,主要用于气阴两虚、烦热口渴、虚劳咳嗽、跌扑损伤、关节疼痛、咳血、外伤出血等症的治疗[1-2]。现代研究表明,珠子参化学成分为皂苷、多糖、氨基酸和多种微量元素[3-7],其中皂苷类成分为珠子参主要活性成分,药理实验表明皂苷类成分具有镇痛镇静、提高免疫力、抗肿瘤等活性[8-11],珠子参的质量控制大多用薄层法鉴别竹节参皂苷Ⅳa和人参皂苷Ro[12]、高效液相色谱法测定竹节参皂苷Ⅳa和人参皂苷Ro[13-15],而对珠子参其他皂苷类成分的文献报道较少,因此,本研究室对珠子参中皂苷成分开展了进一步的研究。
珠子参作为“金品”系列产品痛舒胶囊、痛舒片的重要原料之一[16],药材需求量逐年增加,为此,本研究所开展了珠子参野生资源调查并实施规范化移植繁育试验,最终形成了珠子参规模化种植基地,但由于药材受气候等生态环境的影响,不同批次药材所含化学成分较难控制[17],所以我们运用薄层色谱鉴别及HPLC特征图谱对同一产地不同地块的10批次珠子参药材进行考察,为珠子参药材质量的有效控制奠定了方法基础。
1 仪器及材料
1.1 材料
人参皂苷Ro(批号:111903-201302),竹节参皂苷Ⅳa(批号:111861-201102)均购于中国食品药品检定研究院,虎刺葱木皂苷Ⅵ[18]、竹节参皂苷Ⅳ[19]、竹节参皂苷Ⅰb[19]为本研究室自制。珠子参PanaxjaponicusC.A.Mey.var.major(Burk.) C.Y.Wu et K.M.Feng采自云南丽江种植基地,经云南省药物研究所邱斌高级工程师鉴定为正品,标本保存于云南省药物研究所标本室,共采集了10批珠子参药材,编号为S1~S10。
1.2 试剂
乙腈(色谱级,Merck公司),去离子水(色谱级Milli-Q纯化),其他试剂均为分析纯。
1.3 仪器
UltiMate 3000高效液相色谱仪(LC-20AT四元泵、SIL-20A自动进样仪、CTO-20A柱温箱、SPD-M20A紫外检测器);超声波清洗器SK8200 HP(上海科导超声仪器有限公司);METTLER-TOLEDO AG-285型电子分析天平[METTLER-TOLEDO(上海)有限公司];硅胶G[薄层层析用,Meck化工技术(上海)有限公司]。
2 方法与结果
2.1 薄层色谱研究
2.1.1 药材供试品及对照品溶液的制备 取珠子参药材粉末1.0 g,加甲醇10 mL,超声处理(53 kHZ,400 W)40 min,放冷,过滤,滤液回收溶剂至干,残渣加水20 mL加热使溶解,用水饱和正丁醇振摇提取3次,每次20 mL,合并正丁醇液,回收至干,残渣加甲醇5 mL使溶解,作为供试品溶液。精密称取竹节参皂苷Ⅳa、人参皂苷Ro、虎刺葱木皂苷Ⅵ、竹节参皂苷Ⅳ、竹节参皂苷Ⅰb对照品4.0 mg,分别加入甲醇4 mL超声溶解制成1.0 mg·mL-1对照品溶液。
2.1.2 薄层色谱鉴别 分别吸取供试品溶液、对照品溶液各2 μL,点于同一硅胶G 薄层色谱板上,以正丁醇-乙酸乙酯-甲醇-甲酸-水(5∶10∶1.2∶1.1∶4.5)上层液为展开剂,预饱和15 min展开,展距10 cm,取出晾干,以10%硫酸乙醇溶液喷雾显色,105 ℃加热至斑点清晰,置365 nm紫外灯下观察,见图1。

注:1.竹节参皂苷Ⅳa;2.竹节参皂苷Ⅳ;3.竹节参皂苷Ⅰb;4.珠子参药材;5.人参皂苷Ro;6.虎刺葱木皂苷Ⅵ。图1 珠子参及对照品TLC图
分别从不同薄层板、不同展距、不同饱和时间、不同温度、不同湿度方面验证珠子参药材的薄层鉴别条件,1)薄层色谱板分别选取青岛海洋硅胶G板、默克硅胶G板、鼎康硅胶G板;2)不同展距:8、10、12 cm;3)不同预饱和时间:0、15、30 min;4)不同温度:4、25、32 ℃;5)不同湿度:18%、58%、72%。以上各条件下的展开效果对比发现,除不同硅胶色谱板与不同饱和时间对珠子参中皂苷类成分的分离度有较明显的影响外,其他条件下影响较小,确定珠子参药材薄层色谱鉴别条件为:吸取珠子参供试品溶液及各对照品溶液2 μL(接触点样)点于默克硅胶G薄层色谱板上,以正丁醇-乙酸乙酯-甲醇-甲酸-水(5∶10∶1.2∶1.1∶4.5)上层为展开剂(15 mL),预饱和薄层色谱板15 min,上行展开,展距10 cm,温度25 ℃,湿度58%,取出,晾干,以10%硫酸乙醇溶液喷雾显色后,105 ℃加热至斑点清晰,在365 nm紫外灯下检视,10批不同批次的珠子参皂苷类成分与混合对照品溶液的薄层色谱见图2。
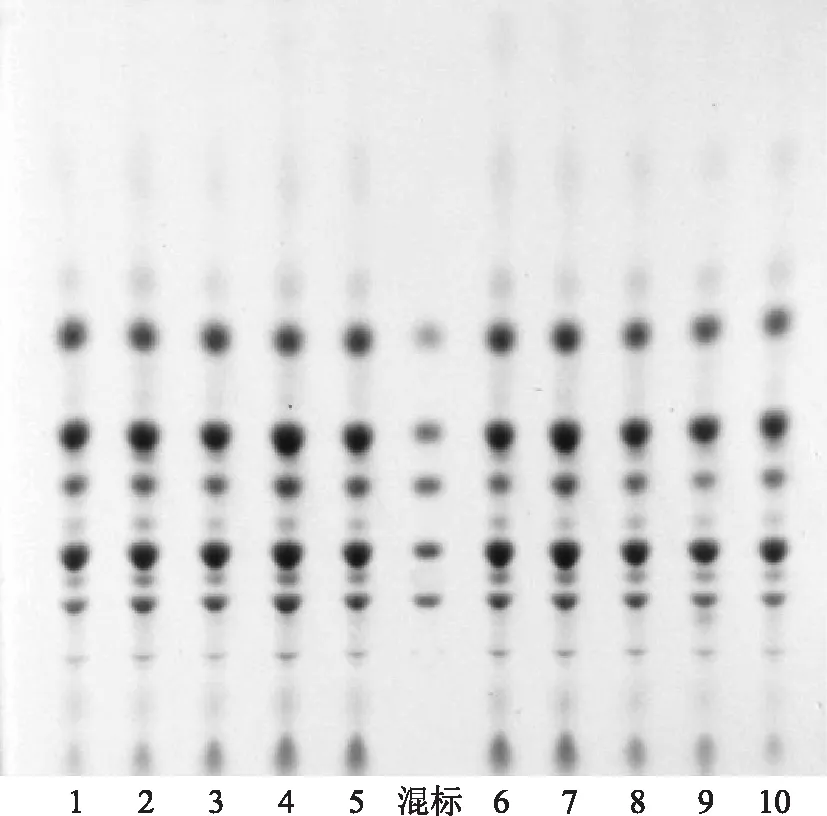
图2 不同批次珠子参药材及混合对照品TLC图
2.2 HPLC特征图谱研究
2.2.1 色谱条件 色谱柱:Waters XTerra MS C18(250 mm×4.6 mm,5 μm);流动相:乙腈(B)-0.2%磷酸水(A);梯度洗脱:0~10 min,25%~30% B;10~20 min,30% B;20~30 min,30%~33% B;30~42 min,33% B;42~45 min,33%~25% B;45~50 min,25% B;检测波长:203 nm;柱温:25 ℃;体积流量:1.0 mL·min-1;进样量:10 μL。
2.2.2 供试品溶液的制备 称取珠子参粉末(过五号筛)约0.1 g,置具塞锥形瓶中,精密加入65%乙醇25 mL,密塞,称定质量,超声处理(53 kHZ,400 W)40 min,取出,放冷,再称定质量,用65%乙醇水溶液补足减失的质量,摇匀,滤过。滤液经0.45 μm微孔滤膜滤过,即得。
2.2.3 对照品溶液的制备 精密称取各对照品适量,分别置于25 mL棕色量瓶中,用适量65%乙醇水溶液溶解并稀释至刻度,分别制成含竹节参皂苷Ⅳa 280.0 μg·mL-1、人参皂苷RO280.5 μg·mL-1、虎刺葱木皂苷Ⅵ 281.1 μg·mL-1、竹节参皂苷Ⅰb 280.7 μg·mL-1、竹节参皂苷Ⅳ 280.6 μg·mL-1的对照品储备液,经0.45 μm微孔滤膜滤过即得。
2.2.4 方法学考察
2.2.4.1 精密度试验 取同一供试品溶液,连续进样6次,检测指纹图谱,计算主要色谱峰的相对保留时间和相对峰面积的RSD,结果表明各色谱峰相对保留时间的RSD<0.2%,相对峰面积的RSD<0.5%,表明本仪器的精密度良好。
2.2.4.2 重复性试验 取同一供试品6份,按2.2.2项下的方法平行制备6份供试品溶液,进样后进行分析,检测指纹图谱,计算主要色谱峰的相对保留时间和相对峰面积的RSD,结果表明各色谱峰相对保留时间的RSD<0.2%,相对峰面积的RSD<1.0%,表明该分析方法重复性良好。
2.2.4.3 稳定性试验 取同一供试品溶液,分别在0、2、4、8、12、24 h进样,检测指纹图谱,计算主要色谱峰的相对保留时间和相对峰面积的RSD,结果表明各色谱峰的相对保留时间的RSD<0.2%,相对峰面积的RSD<2.0%,表明供试品溶液在24 h内稳定。
2.2.5 HPLC特征图谱的建立及相似度评价
2.2.5.1 共有峰指认 将10批同一产地不同地块收集的珠子参药材分别按照2.2.2项制成供试品溶液,按2.2.1项色谱条件进样分析,得到各样品的HPLC特征图谱。同时精密吸取对照品溶液适量,按2.2.1项色谱条件进样10 μL,参照5个对照品的色谱行为及其DAD检测紫外光谱图,在样品色谱图上对其峰进行指认,确认了5个成分,其中1号峰为虎刺葱木皂苷Ⅵ,2号峰为人参皂苷Ro,3号峰为竹节参皂苷Ⅰb,4号峰为竹节参皂苷Ⅳ,5号峰为竹节参皂苷Ⅳa,见图3。
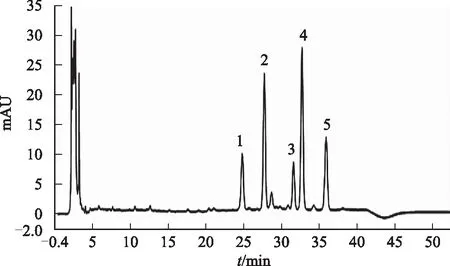
注:1.虎刺葱木皂苷Ⅵ;2.人参皂苷Ro;3.竹节参皂苷Ⅰb;4.竹节参皂苷Ⅳ;5.竹节参皂苷Ⅳa。图3 珠子参HPLC图
2.2.5.2 相似度计算 将所得指纹图谱导入“中药色谱指纹图谱相似度评价系统(2004A)”,计算各样品指纹图谱(见图4)与生成的对照图谱的相似性系数,结果见表1。从各批药材的HPLC 特征图谱与相似性系数都可以看出,不同批次珠子参药材特征图谱相似度均大于0.95,表明同一产地不同地块的珠子参药材的化学组成十分相似。
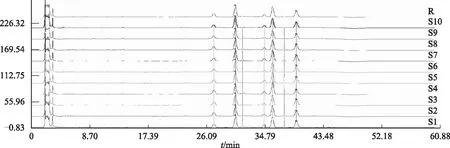
注:R.对照;S1~S10.批次。图4 不同批次珠子参药材HPLC特征图谱
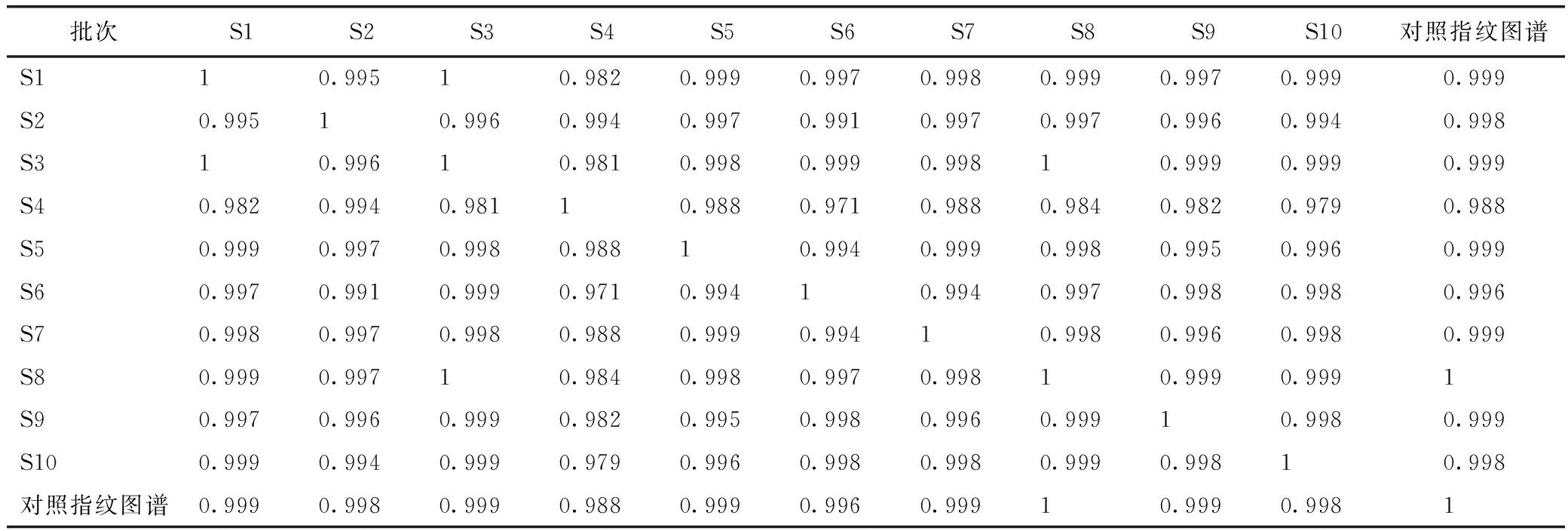
表1 10批珠子参药材的相似度结果
3 讨论
3.1 薄层色谱
该法与《中华人民共和国药典》2015年版一部珠子参项下的薄层色谱鉴别方法相比较,能更加客观、准确、全面地反映珠子参药材的质量。先前相关文献主要通过人参皂苷Ro、竹节参皂苷Ⅳa对珠子参药材进行鉴别,忽略了其余3个主要皂苷成分,不能全面地评价珠子参药材的质量,本实验对珠子参药材的薄层鉴别方法进行了完善,对进一步评价珠子参药材的质量具有一定指导意义。
3.2 HPLC特征图谱色谱条件的优化
尝试多种流动相体系,甲醇-水,乙腈-水,乙腈-磷酸水体系,结果表明,乙腈-0.2%磷酸水体系分离的效果较好,分析时采用梯度洗脱,分析条件如2.2.1项下所示,峰分离度较好,保留时间适中。
本实验建立了珠子参药材的HPLC指纹图谱,相对于已有的图谱分离度更好、指纹性更强,且对5个共有性指标成分进行了指认,该方法简便、快速,可用于珠子参药材的质量控制,为实现珠子参药材规范化种植及质量的均一、稳定奠定了方法基础。
[1] 国家药典委员会.中华人民共和国药典:一部[S].北京:中国医药科技出版社,2015:271-272.
[2] 国家中医药管理局《中华本草》编委会.中华本草:第五册[M].上海:科学技术出版社,1999:835-838.
[3] 何瑞,刘琦,刘银环,等.珠子参叶化学成分研究[J].中国中药杂志,2014,39(9):1635-1638.
[4] 赵东东,宋小妹,汤海峰,等.珠子参叶的皂苷成分研究[J].中南药学,2013,11(2):85-88.
[5] 施丽娜,詹尔益,张玉珠,等.珠子参挥发油的化学成分[J].昆明医学院学报,1992,13(1):46-48.
[6] 池群,郭建文.珠子参的微量元素分析比较[J].西北药学杂志,1993,8(2):61-64.
[7] 杨芳,杨万林,陈锦玉,等.珠子参地上部分氨基酸测定及营养评价[J].氨基酸和生物资源,2013,35(2):1-4.
[8] 考玉萍,姜炜,宋小妹.珠子参叶总皂苷抗疲劳抗应激作用的实验研究[J].陕西中医,2008,29(8):1092-1094.
[9] 陈涛,陈茂华,胡月琴,等.珠子参多糖抗肝癌作用的实验研究[J].时珍国医国药,2010,21(6):1329-1331.
[10] 胡卫,陈涛.珠子参抑制小鼠肝癌及诱导细胞凋亡的研究[J].时珍国医国药,2005,3(10):3-5.
[11] 李巧云,赵恒,岳松健,等.大叶珠子参总皂甙的镇痛镇静作用研究[J].华西药学杂志,1993,8(2):90-92.
[12] 刘越,张旋,贾璞,等.珠子参薄层色谱鉴别方法的研究[J].西北药学杂志,2010,25(3):182-184.
[13] 李渊源,王青,王薇,等.不同采收期珠子参中竹节参皂苷Ⅳa含量测定[J].陕西中医学院学报,2012,35(3):74-75.
[14] 曹红云,牛延菲,罗宗斌,等.不同炮制方法对珠子参中竹节参皂苷Ⅳa含量的影响[J].南京中医药大学学报,2010,26(2):143-145.
[15] 宋小妹,李伟东,房方,等.珠子参药材质量标准研究[J].南京中医药大学学报,2010,26(2):143-145.
[16] 云南省药物研究所.云南省药物研究所制药厂金品系列药品临床推介[J].云南中医中药杂志,2004,25(5):50.
[17] 张志清,曹蕾,宋亮,等.珠子参类药物历史沿革与应用展望[J].云南中医中药杂志,2012,33(3):68-71.
[18] 方乍浦,雷江凌,曾宪仪.虎刺楤木根皮化学成分研究[J].植物学报,1995,37(1):74-80.
[19] Chan H H,Hwang T L,Sun H D,et al.Bioactive constituents from the roots ofPanaxjaponicasvar.majorand development of a LC-MS/MS method for distinguishing between natural and artifactual compounds [J].J Nat Prod,2011,74(4):796-802.
StudyonTLCandHPLCFingerprintsofSaponinConstituentsfromtheRootsofPanaxjaponicusvar.major
ZHANGHaiyuan,XIAWeijun,XIEJiaying,TANGMengyun,ZHANGYingjie,CUIRong,MEIShuangxi,YANGLiguo*
(YunnanInstituteofMateriaMedica,YunnanBaiYaoGroupInnovationandR&DCenter,YunnanProvinceCompanyKeyLaboratoryforTCMandEthnicDrugofNewDrugCreation,Kunming650111,China)
Objective:To establish and improve the TLC and HPLC fingerprint methods for identification of the roots ofPanaxjaponicusvar.major.Methods:The TLC method was adopted to distinguish saponin constituents of the roots ofP.japonicusvar.major.A separation was performed on a Waters XTerra MS C18chromatographic column(250 mm×4.6 mm,5 μm)with gradient elution.The mobile phase consisted of acetonitrile-phosphoric acid water solution(0.2%).The analysis time was 55 min and the detection wavelength was 203 nm.Results:The TLC separation degree was good,the separated spots were clear with specific attribute,and the HPLC fingerprints of saponin constituents of the roots ofP.japonicusvar.majorwas set up.Conclusion:The TLC and HPLC fingerprint methods are stable,accurate,reliable,and can provide a scientific basis for the quality control of the roots ofP.japonicusvar.major.
The roots ofPanaxjaponicusvar.major;saponins;TLC;HPLC fingerprints
2016-06-16)
云南省科技领军人才培养计划项目(2014HA001)
*
杨立国,工程师,博士,研究方向:天然药物化学;E-mail:yangliguo5366@163.com
10.13313/j.issn.1673-4890.2017.3.011
