一种依泽替米贝的合成方法
2017-09-20韩勇
韩勇
海南海灵化学制药有限公司
一种依泽替米贝的合成方法
韩勇
海南海灵化学制药有限公司
化合物E-2与特戊酰氯反应后再与S-(+)-4-苯基-2-噁唑烷酮反应生成化合物E-3,E-3与甲基噁唑硼烷还原剂还原得到E-4,E-4在钛化合物催化下与化合物E-1反应得到E-5,然后环合脱保护得到目标化合物依泽替米贝。该合成路线段,反应条件温和,工艺操作简单,成本较低,产品收率和纯度较高,适合工业化生产。
选择性胆固醇吸收抑制剂;依泽替米贝;合成;工艺
依泽替米贝(1-(4-氟苯基)-(3R)-[3-(4-氟苯基)-(3S)-羟基丙基]-(4S)-(4-羟基苯基)-2-丙内酰胺)又称依折麦布、依替米贝,是由先灵葆雅公司和默克公司共同研制开发的首个选择性胆固醇吸收抑制剂,本品是第一个获得美国FDA批准上市的胆固醇吸收选择性抑制剂类药物。商品名“益适纯”,“EZETROL”。
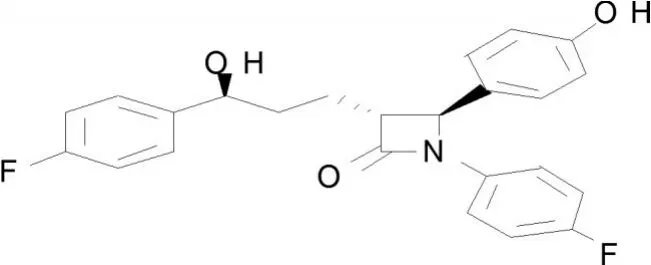
其合成路线如下:
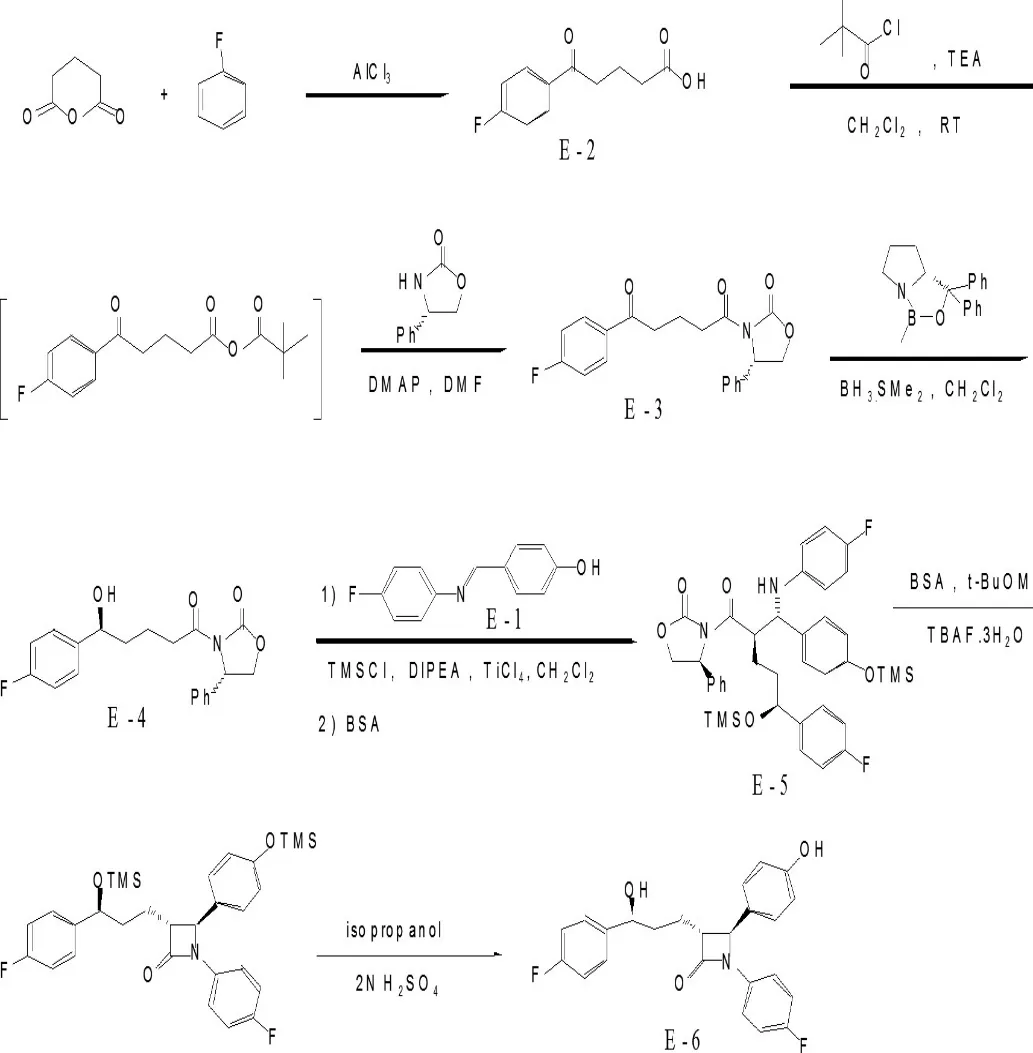
1、实验部分
1.1、E-1的合成

在250mL三颈瓶中加入14.4g(118mmol)对羟基苯甲醛,50mL异丙醇,加热至50℃,搅拌15min,原料未溶解完全,于此温下缓慢滴加11.4mL(120mmol)对氟苯胺,约20min滴完。滴完继续保温反应2h。冷至室温再搅拌30min,过滤,用15mL异丙醇洗涤虑饼,真空干燥,得浅黄色固体20.3g,Y=80%,M.P:180-181℃,文献值180-182℃。
1.2、E-2的合成
在1L三颈瓶中加入205.9(1.54mol)gAlCl3,500mL二氯甲烷,搅拌下于0℃加入80g(0.7mol)戊二酸酐的125mL二氯甲烷溶液,体系搅拌30min,缓慢滴加67.4g(0.7mol)氟苯,0℃下搅拌反应5h(TLC显示反应完全)。然后将反应液倒入2000mL冰水中,过滤,水洗滤饼,真空50℃干燥,得110g产物,收率Y=75%。
1.3、E-3的合成
将10.5g(0.05mol)E-2、50mL二氯甲烷加入250mL反应瓶中,加入13mL(0.093mol)三乙胺,于室温搅拌下缓慢滴加6mL (0.0487mol)特戊酰氯,约20min滴完,继续室温搅拌反应2h至TLC显示反应基本结束,加入5.55g(0.034mol)恶唑烷酮、0.9(0.0073mol) gDMAP,加热回流反应8h,TLC显示反应完全,体系冷至室温,搅拌下倒入至40ml2N硫酸中,再搅拌30min,静置分层,有机层用40ml5%碳酸氢钠洗涤,浓缩,得一油状物,加入60ml异丙醇结晶得类白色产物,干燥得10.4g。
1.4、E-4的合成
在100ml反应瓶中加入5ml二氯甲烷,2M的硼烷二甲硫醚的四氢呋喃溶液7.5ml,冷至-5~0℃,加入0.75ml 1M的甲基噁唑硼烷的甲苯溶液,搅拌15min,保持内温-5~0℃缓慢滴加5gE-3的20ml二氯甲烷溶液,约3h左右滴完。滴完继续保温搅拌反应2h,TLC显示反应完全。缓慢滴加2ml甲醇,产生大量气泡,10ml5%双氧水,1ml4N硫酸,搅拌15min。静置分层,有机层分别用10ml 2N硫酸、25ml 5%Na2SO3、25ml 10%盐水洗涤,无水硫酸镁干燥,过滤,浓缩,得5.1g淡黄色稠状油状物。收率Y=100%,直接用于下步环合反应。
1.5、E-5的合成
N2保护下将上步所得E-4二氯甲烷溶液、5gE-1加入反应瓶中,补加二氯甲烷至75mL,冷至-10℃,缓慢滴加13ml N,N-二甲基乙胺,保持体系温度<-5℃,得一悬浊液,E-1未溶解完全。接着控温缓慢<-5℃滴加7ml三甲基氯硅烷,体系逐渐溶清,得一淡黄色溶液。继续保温搅拌反应2h,TLC显示硅烷化反应基本完成,降低体系温度至-25~-30℃,缓慢滴加1.8ml四氯化钛的10ml二氯甲烷溶液,滴毕继续搅拌反应4h,体系颜色逐渐加深,最后得一深红色溶液。反应完毕缓慢滴加4ml冰醋酸,搅拌下将反应液倒入70ml的7%酒石酸水液中,继续搅拌2h,体系温度缓慢升至室温。再加入25ml 10%亚硫酸氢钠水溶液,再继续搅拌2h,有刺激性气体产生,固体逐渐溶解,得一黄褐色体系。静置,分层,,有机相水洗,干燥,浓缩,加入4.3mlBSA回流1h,得一红褐色溶液,蒸出溶剂,加入10ml乙酸乙酯和20ml石油醚结晶,干燥,得类白色固体6.5g,收率Y=65%。
1.6、E-6(依泽替米贝)的合成
将6gE-5与60ml乙腈混合,得悬浊液,搅拌下加入3.8ml BSA、0.01g四丁基氟化铵,体系迅速溶清,室温搅拌反应2h,TLC显示反应完全,加入0.35ml冰醋酸,搅拌5min后浓缩蒸干溶剂,得一褐色粘稠状液体,加入25ml异丙醇和2.5ml 2N硫酸的混合液,室温搅拌反应1h,硅烷基水解,TLC显示反应完全后缓慢滴加45ml水,体系逐渐析出类白色固体。滴加完毕搅拌15min后过滤,水洗,干燥,得3.3g产物,Y=91.5%。
2、结果与讨论
E-1亚胺吸潮易水解,保存注意干燥;
E-2与特戊酰氯反应进行缓慢,应尽量使E-2反应完全;
E-4蒸馏时温度不宜过高,防止消旋;
E-5合成时加入醋酸放热严重,注意控制滴加速度;
总之,本工艺具有经济、高效、环保、反应操作简单的特点,适合工业生产。
[1]专利WO00/34240.
[2]John W Clader,The Discovery of Ezetimibe:A View from Out⁃side the Receptor[J].Med Chem,2004,47(1).
[3]黄伟,岑均达.Ezetimibe的合成[J].中国医药工业杂志,2006, 37(6).
