基于COI基因对混养缅甸蟒个体来源及遗传多样性分析
2017-09-19樊翠平段玉宝田秀华白素英
樊翠平段玉宝田秀华白素英
(1.东北林业大学野生动物资源学院,哈尔滨,150040;2.西南林业大学林学院,云南省森林灾害预警与控制重点实验室,昆明,650224)
基于COI基因对混养缅甸蟒个体来源及遗传多样性分析
樊翠平段玉宝田秀华1*白素英1
(1.东北林业大学野生动物资源学院,哈尔滨,150040;2.西南林业大学林学院,云南省森林灾害预警与控制重点实验室,昆明,650224)
稿件运行过程
缅甸蟒; 个体来源; COI; 遗传多样性; 遗传距离
缅甸蟒(Pythonbivittatus)是世界上最巨型的6种无毒蛇类之一[1],隶属于蛇亚目(Serpentes)、蟒科(Pythonidae),被列为 《中国物种红色名录》极危物种,为国家Ⅰ级和 CITES附录Ⅱ保护动物[2]。缅甸蟒在中国分布于福建(南平、金门)、广东、香港、海南、广西、云南、贵州(望谟、罗甸、紫云)、西藏(墨脱)、江西、四川(内江)等地[1-4]。
分子系统学是通过分子技术,从分子水平研究生物分类和生物进化关系的一门学科[5],这项技术需要大量特定标记的遗传数据,通过对遗传数据进行统计整理,分析出个体间的遗传差异,达到对结果进行解释的目的。分子系统学的高速发展,使分子标记被运用在生物学领域研究的方方面面[6]。线粒体(mitochondrial)是存在于真核生物细胞内的一种可以进行有氧呼吸的细胞器,同时也是动物体细胞质遗传信息的唯一载体[7]。线粒体遗传是严格的细胞质母系遗传,它具有分子质量小,结构简单,不易发生插入和缺失等特点,是研究低级分类单元较好的分子标记方式,在多种动物类群的系统进化研究中得到广泛应用[8]。细胞色素c氧化酶亚单元I(Cy-tochrome oxidase subunitⅠ,COI)基因是线粒体蛋白质编码基因之一[3],该基因序列虽然进化速率较低,序列相对保守,但却也能够发生充足的基因突变,提供丰富的DNA信息[9-10],被认为是一种非常好的分子生物学标记基因,这也是COI DNA条形编码基因常被选用作为研究基因的主要原因[4]。海内外学者通过对线粒体控制区基因分析成功判定缅甸蟒属于本土物种,通过对微卫星标记分析缅甸蟒遗传多样性。台湾师范大学教授You等利用线粒体Cytb和COI对33条来自台湾金门、福州动物园以及越南的缅甸蟒进行了分子系统发育研究,单倍型网络显示了金门种群与大陆种群亲缘关系较近,证实了金门岛的缅甸蟒并非外来种,是台湾首例被列为本土种的蟒蛇[11];刘雅芳等对海南和越南2个已知来源的缅甸蟒种群,基于控制区II序列进行建树、并计算两群体的固定指数(Fst),得出海南缅甸蟒与越南缅甸蟒已经出现明显分化的结果[12]。段玉宝等人通过微卫星标记得出缅甸蟒海南养殖群体在近期没有经历过瓶颈效应,群体数量没有出现过明显下降[10]。杨宝山等通过COI基因序列变异对不同地理种群的银杏大蚕蛾(DictyoplcajaponicaButler)构建系统发育树,分析结果表明南方组和北方组种群间已经按地理位置形成了一定的地理格局,AMOVA分析显示北方组和南方组之间已经具有明显的遗传分化,因此可以根据COI基因序列变异将这13个地理种群划分为北方和南方2个组的进化分枝[13]。张迪等人在基于COI基因序列的太湖新银鱼(Neosalanxtaihuensis)遗传多样性中认为线粒体COI基因非常适合银鱼科(Salangidae)鱼类的物种鉴别和系统发育研究,在同种不同地理种群间遗传关系研究中也同样适宜,但若结合Cytb等基因一并分析,可以获取更多的遗传信息[14]。郑文娟等人基于线粒体COI基因序列探讨泥蚶(Tegillarcagranosa)的遗传分化,得出我国沿海地区7个泥蚶群体已经分化形成了二大类群:福建以北(包括福建)的5个群体(江苏盐城、浙江奉化、浙江乐清养殖和自然群体、福建福鼎)形成一个类群,福建以南的2个群体(广东湛江、海南海口)形成一个类群[15]。You基于Cytb和COI基因序列的蟒蛇分子鉴定中,表明福州动物园的蟒蛇是福建本地种群和东南亚种群混养的,其中金门种群和Fuzhou1和Fuzhou4组成福州本地种群,Fuzhou2、Fuzhou3、Fuzhou5与海南缅甸蟒蛇及越南蟒蛇等组成东南亚种群。该文章表明金门蟒蛇非来源东南亚,与金门原生种群与福建本地种同源[11]。
近些年,中国境内蟒蛇野外数量急剧减少,缅甸蟒人工养殖的成功繁育和驯化,为野外种群的扩增提供了可能。而国内对于该物种原人工繁育种群的遗传学、分子生物学等相关研究十分缺乏。海南东盛弘蟒业科技股份有限公司饲养的缅甸蟒其产地与来源不同,由于没有规范的来源记录,且不同地理种群的蟒蛇在外形特征上不易区分,以至不能准确鉴定。为了更好地对蟒蛇进行科学的鉴别和保护,本研究首次对人工混养养殖种群基于COI基因序列进行了遗传多样性、遗传距离较为详尽研究,并重新确定人工混养养殖种群的物种来源,为人工繁育的缅甸蟒种群健康发展和野生资源的迁地保护提供了分子生物学参考。
1 材料与方法
1.1实验材料
在海南东盛弘蟒业科技股份有限公司,采用无损伤取样法收集缅甸蟒蛇蜕样本,共采集28条缅甸蟒刚刚褪去的蛇蜕,冰冻带回实验室,-80℃保存。
1.2实验方法
1.2.1基因组DNA的提取
利用经典的苯酚-氯仿方法提取缅甸蟒的蛇蜕基因组DNA:缅甸蟒基因组DNA通过酚、氯仿多次抽提,后用无水乙醇和70%的乙醇沉淀DNA;室温干燥后加50 μL TE溶液溶解,-4℃保存或-20℃储存。用紫外分光光度法检测DNA的浓度,然后稀释成50 ng备用。
1.2.2引物的扩增
本研究使用You等[11]报道的COI引物,引物由博仕生物技术股份有限公司合成。
F:5′-CCCTTATGAGTAGATTTACAGCCTA-3′
R:5′-GGATTGGGGCGTACATATTGTTTAGT-3′
PCR扩增的目的片段为1 500 bp,扩增体系为50 μL,其配比为:8×PCR Buffer 5 μL,dNTP Mixture 5 μL,rTaq酶0.5 μL,Primer 1和Primer 2均是1 μL(最终浓度),模板5~8 μL,然后补增灭菌的去离子水至50 μL。
在Eppendorf梯度PCR仪上进行PCR扩增,PCR反应条件:依次预变性温度95℃时长5 min,变性温度95℃时长28 s,退火温度55℃时长28 s,延伸温度72℃时长88 s,35个循环后72℃延伸8 min,4℃保存。将PCR扩增产物在浓度为1%的琼脂糖凝胶中进行电泳检测后,采用爱思进生物技术(杭州)有限公司的AxyPrep DNA凝胶回收试剂盒对PCR产物进行纯化回收,纯化回收后的样品送博仕生物公司使用ABI3728全自动测序仪进行双向测序。
1.2.3数据处理
将测得的基因序列,用Seqman软件进行拼接,人工进行峰图矫正后,使用MEGA 5.05与Genbank中的基因序列进行比对、剪切;使用DnaSP 5.8.01软件统计多态位点数、核苷酸多样性(Pi)、单倍型多样性(Hd)和核苷酸平均差异数(K),确定单倍型。在NCBI中下载已知的缅甸蟒COI序列,用MEGA 5.05采用最大似然法(Maximum Likelihood,ML),以 Modeltest 3.7中h LRT筛选出的最优模型GTR+I+G 在 PAUP*4.0 b8中构建ML树。用MEGA 5.05计算不同种间的遗传距离。
表1 GenBank下载序列详情
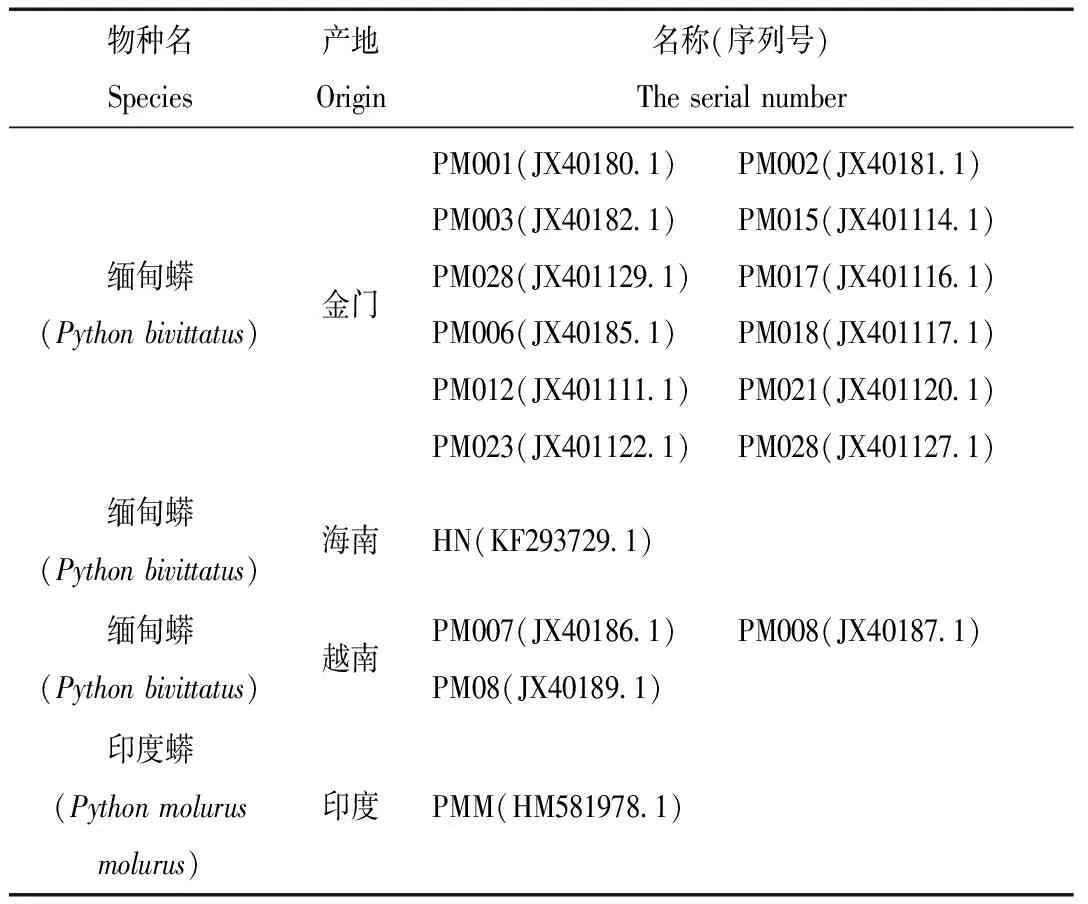
Tab.1 GenBank download sequence for details
2 结果
2.1 COI序列基因片段特征及遗传多样性
测序得到的28条缅甸蟒mtDNA COI基因序列,经过比对剪切得到COI基因全序列,片段长度为1 428 bp。其中T、C、A和G的含量分别为29.8%、15.5%、27.9%和26.9%,A+T的含量(54.8%)高于C+G的含量(45.2%)。保守位点(C)占89.6%,变异位点(V)占9.6%,简约信息位点(Pi)占1.7%。缅甸蟒混养种群存在的单倍型数为8个,单倍型多样性(Hd)为(0.726±0.068),核苷酸多样性Pi为(0.003 57±0.000 56),平均核苷酸差异数k是5.099。
2.2 系统发育树的构建
在GenBank分别下载3条越南缅甸蟒、1条海南缅甸蟒和12条金门缅甸蟒与1条印度蟒(表1)的COI基因序列,与本研究所检测的28条养殖缅甸蟒样本的COI同源基因序列采用邻接法构建ML(图1)。
由图1的ML树拓扑结构可以看出,12条金门缅甸蟒聚成一小支,与8条养殖缅甸蟒与海南缅甸蟒HN(KF293729.1)聚在一起,和其余20条养殖缅甸蟒聚与3条越南缅甸蟒(PM008(JX40187.1)、PM007(JX40186.1)、PM08(JX40189.1))聚成的一支共同形成姐妹支,然后与印度蟒(PMM(HM581978.1)聚在一起。

图1 基于COI基因序列构建的全部个体ML系统进化树Fig.1 Phylogenetic tree based on COI gene sequences of all individuals
2.3 遗传距离
用MEGA 5.05计算不同种间的遗传距离,首先对单个个体间遗传距离进行分析,发现全部缅甸蟒个体间遗传距离在0.000 7~0.012 75之间;缅甸蟒个体与印度蟒之间的遗传距离为0.069 02~0.073 80。又根据已知地理分布和系统发育树所显示的差异将样本分为4大类群,分别为海南类群(HN:包括海南个体与8条养殖个体)、金门类群(JM)、养殖群体1(YZ1:包括其余20条聚在一起的养殖个体)和越南群体(YN)。计算4个群体间遗传距离(表2),根据其遗传距离远近判断其归属。结果发现4个缅甸蟒群体内的遗传距离都很接近(0.0~0.002 95)。4个群体间遗传距离在0.002 11~0.006 99之间。其中YZ1群体与YN群体间遗传距离为0.002 52,与海南群体间遗传距离为0.006 99。
表2 基于COI基因序列计算的4个越南缅甸蟒群体间的遗传距离
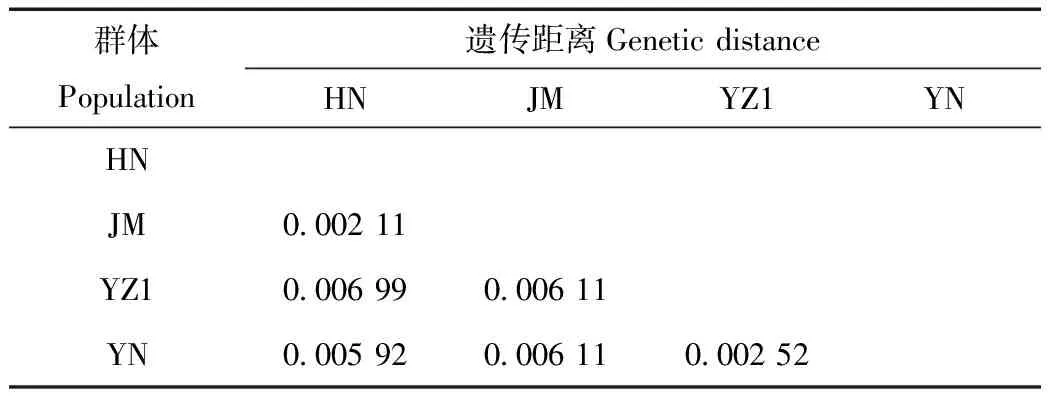
Tab.2 Genetic distance of partial COI gene between four Python bivittatus populations
3 讨论
3.1 COI序列的多态性分析
通过对COI碱基序列的分析可以发现,C碱基在各碱基中所占的比例要低于其他碱基,其中A+T的含量(54.8%)明显高于C+T的含量(45.2%),说明COI的碱基组成具有偏向性,这与脊椎动物线粒体DNA轻链(light strand)COI区核苷酸的碱基组成特点相一致[15]。已有研究表明,线粒体不同基因的进化速率存在差异。本文中检测的28条养殖缅甸蟒COI基因中保守位点(C)占86.9%,变异位点(V)占4.6%,证明COI基因序列相对保守,变异速率慢。COI基因是线粒体13种编码基因之一,因此其进化速率较慢,这与其他动物相关序列的研究结果相同。
人工混养养殖缅甸蟒群体单倍型多样性相对比较高(>0.7),核苷酸多样性较低(<0.007),这表明该群体遗传多样性并不高。已有的研究通过对线粒体DNA序列的遗传变异进行分析,通过单倍型多样性与核苷酸多样性的高低组合将其分为4种类型:第一类是前者较低(< 0.5)同时后者也低(< 0.005);第二类是前者较高但后者较低;第三类是前者较低但后者却较高;第四类是前者与后者同时相对较高[16]。本研究中养殖缅甸蟒的结果属于第二种类型。物种种群数量大和生长环境的不均性是维持自然种群内单倍型多样性较高的基础,种群数量的增长速度也是影响遗传多样性的重要因素[17]。刘雅芳等对缅甸蟒线粒体控制区遗传多样性的研究结果显示缅甸蟒总体上群体遗传特征与本文一致[12]。一个种群能否健康持续的发展,遗传多样性是关键,它关乎着整个种群的长期进化和繁衍,这一点对于人工混养缅甸种群的人工驯养及繁殖也同样重要[18]。遗传多样性低,同样会为人工驯养带来阻碍,因此在人工养殖过程中,我们更应加强遗传管理,明确物种内种群的划分关系,并建立明确的谱系关系,防止近亲衰退。
3.2 系统发育树分析
刘雅芳等人通过对海南和越南已知的缅甸蟒种群的线粒体控制区基因建树并计算两群体间的固定指数Fst,证明海南与越南缅甸蟒在分子水平上已出现明显的分化[12]。本文基于COI核苷酸序列构建的系统发生树显示,缅甸蟒与外群完全分开,ML法系统发育树基部分为2支:由金门缅甸蟒种群与8条养殖蟒蛇个体和海南缅甸蟒个体聚在一起形成1支,3条越南缅甸蟒与其余20条养殖缅甸蟒个体聚在一起形成1支。基于以上建树方法所产生的结果,28条养殖缅甸蟒中有8条缅甸蟒判断其来源地为海南,另外20条缅甸蟒的来源地为越南。由此看出基于COI氨基酸序列重建的系统发育树能够用于缅甸蟒种源划分。
3.3 种群遗传距离分析
Avise认为同一物种的个体间遗传距离一般会保持在0.05之内,当差异超过0.06,该个体已经与其他个体之间发生了明显的亚种或者种的分化[19]。从本研究的结果显示,海南、越南、金门与养殖个体的个体间的遗传距离在0.000 7~0.012 75之间,证明养殖个体与其他野生群体间未发生明显亚种或种的分化。同时做群体间遗传距离,发现YZ1群间与越南群体间的遗传距离为0.002 52,与HN缅甸蟒群体间遗传距离为0.006 99,与JM缅甸蟒群体间遗传距离为0.006 11。YZ1群体与越南缅甸蟒之间遗传距离最近,这也与ML系统发育树结果一致,从而进一步证明了YZ1群体来自越南的结果的准确性。HN群体与JM群体间的遗传距离较近(0.002 11),这也与其物种来源的地理位置远近和系统发育树结果一致。
4 结论
本研究首次通过对28条人工混养养殖缅甸蟒COI基因序列进行扩增,成功准确判断了混养个体的个体来源,28条养殖缅甸蟒样品中有8条来自海南,20条来自越南。养殖场可根据本研究的结果,在以后的种群选育过程中根据不同的繁殖需求选取不同个体进行配对繁殖,同时,根据不同群体建立谱系管理制度,有效避免近交衰退,从而保持缅甸蟒群体内较丰富的遗传多样性。本研究成果为人工繁育的缅甸蟒种群健康发展提供了分子生物学参考,同时对保护蟒蛇资源、人工饲养的种群管理以及明确市场上的产品来源等方面将具有重要的理论参考价值和实际应用价值。
[1] 银星果.蟒蛇幼体对人工饲料的消化率及其生长发育规律研究[D].南宁:广西大学,2014.
[2] 陈英苗,易兰,段成,等.武汉市蛇类市场初步调查[J].资源开发与市场,2010,26(2):132-134.
[3] 张娟,张利平,宗卉,等.家鸭细胞色素C氧化酶Ⅲ(COⅢ)基因的克隆及序列分析[J].中国生物工程杂志,2007,27(3):54-59.
[4] 肖金学,王文强,廉振民.浅议分子生态学的概念[J].延安大学学报:自然科学版,2008,27(1):69-71,75.
[5] 茅云翔,吴菲菲,杜国英.红藻DNA条形码分析技术研究进展[J].中国海洋大学学报:自然科学版,2014,44(8):48-53.
[6] 王利,邢世岩,王正华,等.银杏分子系统学研究进展[J].山东林业科技,2005(6):58-59.
[7] 顾志良,吴常信.牦牛线粒体ND1和ND2基因序列和物种系统发育分析[G]//中国动物遗传育种研究进展—第十三次全国动物遗传育种学术讨论会论文集.北京:中国农业科学技术出版社,2005:33-34.
[8] 潘敏慧,万永继,鲁成,等.用rRNA序列分析家蚕病原性微孢子虫Endoreticulatusbombycis的进化[J].蚕业科学,2003,29(2):196-199.
[9] Barker D G,Barker T M.The distribution of the burmese python,Pythonmolurusbivittatus[J].Bulletin of the Chicago Herpetological Society,2008,43(3):33-38.
[10] 段玉宝,王英树,马建章,等.缅甸蟒养殖种群的遗传多样性分析[J].西南林业大学学报,2016,36(3):163-168.
[11] You C W,Lin Y P,Lai Y H,et al.Return of the pythons:first formal records,with a special note on recovery of the Burmese python in the demilitarized Kinmen islands[J].Zoological Studies,2013,52(1):1-11.
[12] 刘雅芳,侯冠彧,于萍,等.海南与越南缅甸蟒线粒体控制区遗传多样性及差异性分析[J].湖北农业科学,2015,54(2):398-401,408.
[13] 杨宝山,候庆君,王欢,等.不同地理种群银杏大蚕蛾COI基因序列变异与遗传分化[J].昆虫学报,2009,52(4):406-412.
[14] 张迪,雷光春,龚成,等.基于COI基因序列的太湖新银鱼遗传多样性[J].湖泊科学,2012,24(2):229-306.
[15] 郑文娟,朱世华,沈锡权,等.基于线粒体COI基因序列探讨泥蚶的遗传分化[J].动物学研究,2009,30(1):17-23.
[16] 李大命,张彤晴,唐晟凯,等.太湖大银鱼(Protosalanxchinensis)细胞色素b基因序列多态性分析[J].江苏农业学报,2015,31(4):840-845.
[17] Ziegler T,Hendrix R,Vogt M,et al.The diversity of a snake community in a karst forest ecosystem in the central Truon Son,Vietnam,with an identification key[J].Zootaxa,2007,1493:1-40.
[18] 杨博,陈小勇,杨君兴.鱇白鱼线粒体DNA控制区结构和种群遗传多样性分析[J].动物学研究,2008,29(4):379-385.
[19] Avise J C.Molecular markers,natural history and evolution[M].Springer Science & Business Media,2012.
Pythonbivittatus; Individual sources; COI; Genetic diversity; Genetic distance
采用PCR技术首次用缅甸蟒人工养殖混养种群作为实验样本,进行mtDNA COI基因序列扩增,得到28条mtDNA COI基因序列,片段长度为1 428 bp,其中T、C、A和G的含量分别为29.8%、15.5%、27.9%和26.9%,A+T的含量(54.8%)高于C+G的含量(45.2%)。保守位点(C)占89.6%,变异位点(V)占9.6%,简约信息位点(Pi)占1.7%。根据个体系统发育进化树与群体间遗传距离重新判定缅甸蟒人工养殖混养养殖种群的个体来源,并确定28条养殖个体中8条来自海南,20条来自越南。从遗传距离结果可以看出,缅甸蟒养殖群体与野生缅甸蟒相比,并没有出现明显的遗传分化。
Source of Origin and Genetic Diversity of Polyculture Burmese Python (Python bivittatus)Based on COI Gene
Fan Cuiping1Duan Yubao2Tian Xiuhua1*Bai Suying1
(1.College of Wildlife Resource,Northeast Forestry University,Harbin,150040,China;2.College of Forestry,Southweat Forestry University,Key Laboratory of Forest Disaster Warning and Control of Yunnan Province,Kunming,650224,China)
We obtained 28 Cytochrome oxidase subunitⅠ (COI)gene sequences of polyculture burmese python (Pythonbivittatus)using PCR technology.The length of the COI gene fragment was 1428 bp,of which the content of T,C,A and G is 29.8%,15.5%,27.9%,and 26.9%,respectively.Content of A+T (54.8%)was higher than that of C+G (45.2%).Conservative sites (C)accounted for 89.6%,mutation loci (V)accounted for 9.6%,and simple information site (Pi)accounted for 1.7%.Based on phylogeny trees and genetic distance between individuals,the 28 burmese pythons were considered to be 8 individuals from Hainan Province,China,and 20 individuals from Vietnam.According to genetic distance,there was no obvious genetic differentiation between the domestic and wild populations of burmese python.
樊翠平,女,25岁,硕士研究生;主要从事特种经济动物饲养、分子生物学研究。E-mail:1391577155@qq.com 段玉宝,男,33岁,博士,讲师;主要从事保护生物学、分子生物学研究。E-mail:boyciana@163.com
*通讯作者:田秀华,E-mail:tianxiu-hua@163.com
2016-12-06
Q953
A
修回日期:2016-02-24
发表日期:2017-05-10
2310-1490(2017)02-295-05
