肝硬化及肝细胞癌患者肠道微生态组成和结构的初步分析
2017-09-18周冷潇刘保文高英堂韩海燕
周冷潇, 韩 涛, 刘保文, 高英堂, 韩海燕
(1 天津医科大学三中心临床学院, 天津市第三中心医院 肝内科, 天津市人工细胞重点实验室,天津市肝胆疾病研究所, 天津 300170; 2 天津市河东区口腔医院, 天津 300171)
肝硬化及肝细胞癌患者肠道微生态组成和结构的初步分析
周冷潇1, 韩 涛1, 刘保文1, 高英堂1, 韩海燕2
(1 天津医科大学三中心临床学院, 天津市第三中心医院 肝内科, 天津市人工细胞重点实验室,天津市肝胆疾病研究所, 天津 300170; 2 天津市河东区口腔医院, 天津 300171)
目的比较分析肝硬化和肝硬化基础上发生肝细胞癌(HCC)的患者远端肠道菌群的差异,探讨肝硬化患者发生HCC与肠道菌群变化的关系。方法收集2015年12月-2016年5月天津市第三中心医院肝内科住院的慢性肝病患者35例,包括20例肝硬化患者(肝硬化组)和15例肝硬化合并HCC患者(HCC组)。收集患者粪便标本,提取其肠道细菌总DNA,采用Roche 454测序技术对16S rDNA V3-V6可变区测序,并进行生物信息分析(物种分类、丰度分析及多样性分析)。计量资料组间比较采用t检验,计数资料组间比较采用Mann-WhitneyU检验。结果20个肝硬化患者样品平均OTU为306.50±163.76,15个HCC患者样品平均OTU为357.24±168.85,2组患者OTU及alpha多样性指数相比,差异没有统计学意义(P值均>0.05)。粪便菌群序列包括的菌门主要有放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)和变形菌门(Proteobacteria)。属种定量及组成分析发现,肝硬化组和HCC组患者肠道内多种细菌的相对丰度差异显著,其中肝硬化组肠道菌群与HCC组比较,放线菌门(0.21% vs 0.06%,U=89.000,P=0.043)、双歧杆菌属(0.16% vs 0.04%,U=90.000,P=0.046)和梭菌属(0.13% vs 0.08%,U=90.000,P=0.046)所占比例显著升高,理研菌科(0.58% vs 2.30%,U=82.000,P=0.023)和Christenellaceae科(0.01% vs 0.08%,U=84.500,P=0.028)所占比例显著降低。结论与单纯肝硬化患者相比,肝硬化基础上发生HCC者肠道菌群的组成有显著差异,但这些差异与肝硬化基础上发生HCC之间相互作用的具体机制尚不明确。
肝硬化; 癌, 肝细胞; 肠杆菌科
肝硬化是常见的消化系统疾病之一。肝细胞癌(HCC)是我国常见恶性肿瘤之一,其病死率高居恶性肿瘤第二位,是我国社会及医疗的沉重负担[1]。人体胃肠道是一个代谢和免疫系统,也包含着最复杂的人体微生态系统。近年来已有多个研究[2-4]提示人类肠道菌群与肝硬化的发展密切相关。高通量测序技术又称为第二代测序技术,以数据产出通量高为最大特点,在微生物学中广泛应用。16S rDNA基因是编码原核生物核糖体小亚基的基因,由于其分子大小适中、突变率小,是细菌系统分类学研究中最常用和有效的标志。本研究采用高通量16S rDNA测序技术及生物信息分析方法,对人类肝硬化和HCC肠道菌群结构的差异和特征进行了分析。
1 资料和方法
1.1 研究对象 收集2015年12月-2016年5月天津市第三中心医院肝内科住院的慢性肝病患者35例,根据是否合并HCC分为肝硬化组(20例)和HCC组(15例)。肝硬化诊断符合影像学检查(腹部超声、腹部超声造影、腹部CT)结果,HCC诊断符合影像学检查(腹部增强CT、腹部增强MRI)、实验室检查(血清AFP)和(或)肝穿刺活组织检查结果,诊断标准参考《原发性肝癌诊疗规范(2011年版)》[5]。所有入选的肝硬化合并HCC患者均首次确诊为HCC,在采样之前未进行过射频消融、介入、生物治疗等针对HCC的治疗。所有患者均未合并炎症性肠病等胃肠道疾病、其他感染性疾病及糖尿病等其他代谢相关疾病,采样前2周未使用或服用抗生素、微生态制剂、酸奶等益生菌、类固醇等激素、中草药制剂。
1.2 研究方法
1.2.1 粪便标本的留取及提取细菌总DNA 所有患者的粪便样品在排出后放入无菌标本盒,立即置于0 ℃环境下,24 h内置于-80 ℃环境下,冰冻状态下使用北京艾德莱生物科技有限公司粪便基因组DNA快速提取盒(DN23)提取DNA,步骤如下:(1)细胞的裂解与破碎:取约200 mg粪便加入缓冲液;(2)去除细胞碎片、蛋白质和研磨物;(3)使用常规RNA酶消化方法处理RNA。DNA标本于冷藏条件下送至天津生物芯片技术有限公司进行测序及生物信息分析。
1.2.2 16S rDNA测序 对粪便DNA样本进行纯化后,采用Roche 454测序技术对样本16S rDNA V3-V6可变区高通量测序。
1.3 生物信息分析
1.3.1 物种分类与丰度分析 对原始测序数据处理后获取用于分析的数据,使用QIIME (quantitative insights into microbial ecology)[6]软件对数据进行处理,将相似度在97%以上的序列聚类成一操作分类单位(operational taxonomic unit,OTU),OTU的数量初步说明该份样品的物种丰度;之后在每一OTU中选择一序列与greengenes数据库(13_5)[7]进行比对,完成物种组成和分类分析,可在界、门、纲、目、科、属水平明确每一OTU的成分。
1.3.2 多样性分析 利用QIIME[6]软件计算针对单个样品物种多样性分析的alpha多样性指数,包括chao1指数、simpson指数、shannon指数、gini指数等。

2 结果
2.1 一般资料 35例患者中男25例,女10例,年龄42~81岁,平均(57.80±10.04)岁。肝硬化组中男11例,女9例,年龄(54.80±8.64)岁,肝硬化病因为乙型肝炎7例、丙型肝炎1例、自身免疫性肝病4例、酒精性肝病4例、乙型肝炎合并酒精性肝病2例、非酒精性脂肪性肝病1例、隐源性1例;Child-Pugh分级A级12例、B级7例、C级1例。HCC组中男14例,女1例,年龄(61.80±10.67)岁,肝硬化病因为乙型肝炎8例、丙型肝炎1例、乙型肝炎合并酒精性肝病4例、隐源性2例;Child-Pugh分级A级6例、B级9例。
2.2 多样性分析 35份粪便细菌DNA样品共产生原始测序数据为583 662条,经过对原始序列进行质量、长度等过滤,得到最终可以用于分析的序列为470 041条。如表1所示,20个肝硬化患者样品OTU数量为77~529,平均306.50±163.76;15个HCC患者样品OTU数量为21~718,平均357.24±168.85。2组患者OTU及alpha多样性指数相比,差异没有统计学意义(P值均>0.05)。
按97%相似度对样本进行OTU分类,构建稀有曲线(图1)。如图所示稀有曲线没有变平缓的趋势,表明随着测序条数的增加,仍然出现新的OTU,表明本实验的测序深度未达到平台期。2组患者稀有曲线的增长趋势没有显著差异。
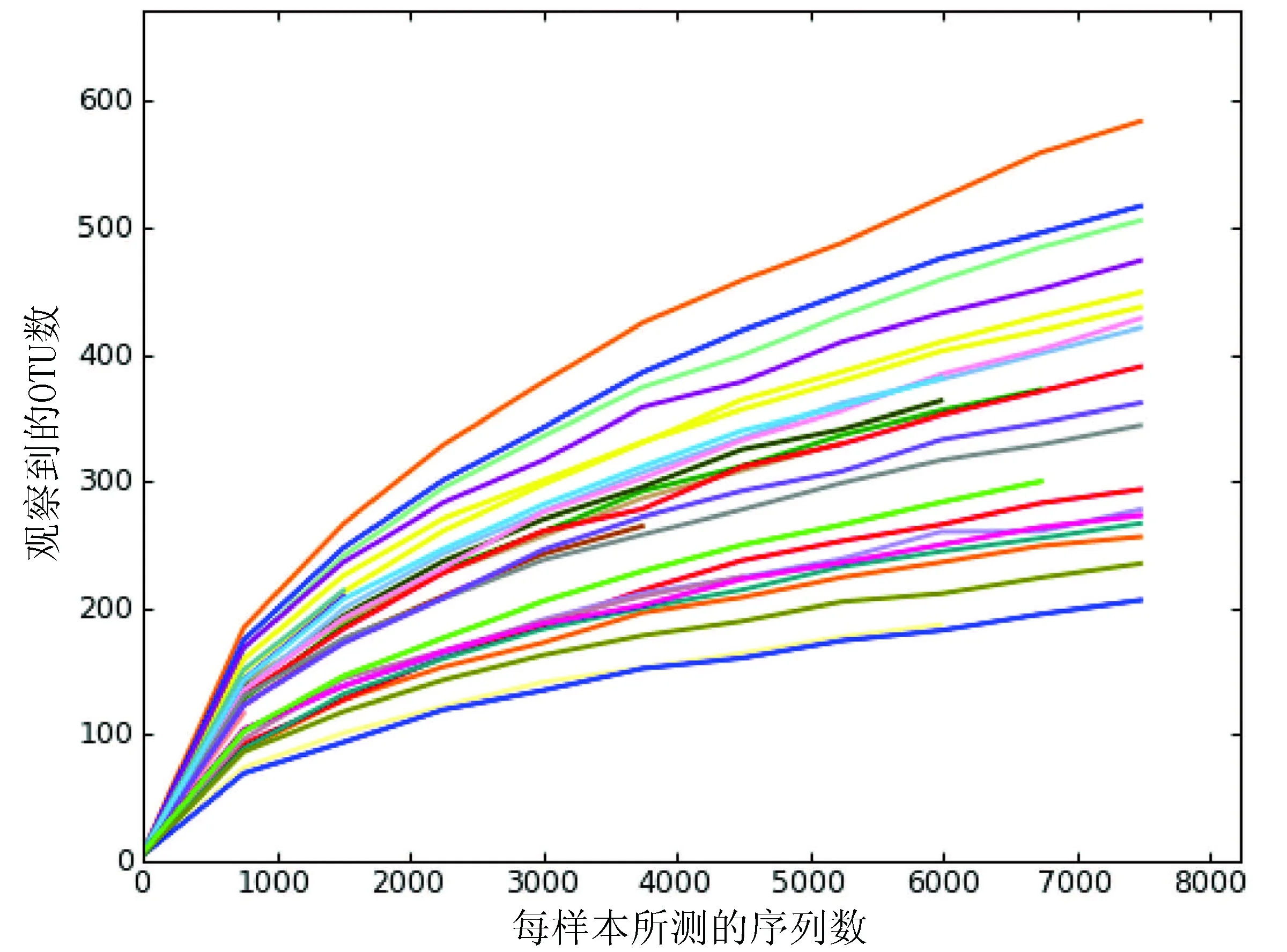
图1 97%相似度的稀有曲线
2.3 主成分分析 对肝硬化患者与HCC患者在门水平上进行细菌主成分分析,发现二者可以较好地区分开(图2)。提示2组患者肠道菌群在门水平上有一定差异。

图2 2组患者肠道菌群门水平上的主成分分析
2.4 序列分类比较
应用rdp分类软件进行序列比对分析,结果显示粪便菌群序列包括的菌门主要有放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)和变形菌门(Proteobacteria),此外部分样品含有极微量的蓝细菌门(Cyanobacteria)、梭杆菌门(Fusobacteria)、互养菌门(Synergistetes)、软壁菌门(Tenericutes)、疣微菌门(Verrucomicrobia)等。
2.4.1 2组患者肠道菌群门水平比较 如表2所示,在门水平上,肝硬化组患者肠道菌群中的放线菌门所占比例比HCC组显著增加(P=0.043);其余几组菌门在2组间所占比例差异均无统计学意义(P值均>0.05)。

表2 2组患者门水平主要细菌平均占比情况(%)
2.4.2 2组患者肠道菌群纲至属水平比较 如表3所示,所列出的细菌在2组患者肠道菌群所占比例差异均有统计学意义(P值均<0.05);其余细菌在2组患者肠道菌群所占比例差异均无统计学意义(P值均>0.05)。
3 讨论
人体胃肠道表面定植的细菌数约为1013~1014[8],包括400种不同种和亚种的细菌[9],肠道包含着最复杂的人体微生态系统,是人体最大的细菌和内毒素池[10]。正常人体肠道屏障包括肠道菌群的正常组成和分布、肠道黏膜屏障以及完整的免疫系统。肝硬化患者胆汁分泌减少[11]和门静脉高压[12]可影响肠道菌群的组成和生长,从而损害肠道屏障,增加肠道通透性,进而导致细菌移位、细菌代谢产物如脂多糖等堆积[13],肠道菌群及其代谢产物穿过肠屏障后可通过门静脉循环进入肝脏。肝Toll样受体(TLR)是一类识别微生物结构保守分子的蛋白质[14],也是脂多糖的受体,在肝脏Kupffer细胞、肝星状细胞和肝细胞内表达。当肝脏持续暴露于肠道菌群代谢产物时,TLR作为脂多糖的靶细胞被激活,参与炎症信号通路从而导致肝细胞反复受损、发生肝纤维化、肝硬化甚至HCC[15]。

表1 2组患者OTU及alpha多样性参数比较

表3 2组患者纲至属水平比较有意义的细菌平均占比情况(%)
在小鼠HCC模型中发现肠道菌群组成和数量的变化导致其代谢产物脱氧胆酸(deocycholic acid,DCA)增多。DCA是一种已知的DNA损伤物,这种损伤物和肥胖诱导的革兰阳性菌成分磷脂壁酸协同作用,可诱发肝星状细胞老化相关分泌表型(senescence-associated secretory phenotype,SASP),通过TLR2途径上调SASP因子和环氧合酶2的表达,而环氧合酶2诱导的前列腺素E2通过前列腺素E型受体4抑制抗肿瘤免疫,从而导致HCC的发生[16],DCA-SASP轴在HCC的发病机制中起重要作用[17];也有研究[18]发现,小鼠体内胆酸蓄积,高水平胆酸的持续刺激导致肝脏发生慢性损伤和炎症反应,在损伤修复过程中,或可出现肝脏异常增殖,最终导致HCC的发生;且在人类非酒精性脂肪肝相关性HCC的发生中也发现了类似的途径[19]。此外,在小鼠HCC模型中,与普通小鼠相比,经过抗生素肠道除菌处理和在无菌条件下生长的小鼠肿瘤数量较少、体积较小[20-21],表明杀灭肠道菌群的生长可抑制肥胖相关HCC,益生菌治疗可减缓肝脏肿瘤生长[22]也支持这一理论。但肠道菌群对肿瘤发生起始阶段的作用尚没有确切结论。
本研究发现HCC患者肠道菌群中放线菌门占比显著低于肝硬化患者,其中以双歧杆菌属为代表,这也与之前的研究结论一致[22]。双歧杆菌是人体肠道中的一组重要细菌,早在20世纪90年代就认为人体肠道中双歧杆菌的减少或消失是“不健康”的[23],现在认为双歧杆菌可在多个方面促进人体健康,包括抑制有害菌生长从而维持肠道微生态平衡、促进消化、调节免疫功能、有助于防止感染等。此外,已有诸多证据显示补充双歧杆菌对慢性肝病患者有益[24-25],而对肝硬化患者补充双歧杆菌是否有预防HCC的作用尚待进一步研究。
既往针对拟杆菌门与HCC相关性的研究相对缺乏, Zhang等[26]研究发现与对照组相比,暴露于二甲基甲酰胺的大鼠模型肠道内理研菌科细菌丰度明显偏高。二甲基甲酰胺有肝毒性,可导致肝脏炎症、纤维化、肝硬化乃至于HCC的发生[27-28]。此外,Liu等[29]认为理研菌科可产生脂多糖。本研究发现HCC组肠道内理研菌科所占比例较肝硬化组明显偏高(P<0.05),但由于HCC的形成机制复杂且不十分明确,现无法确认理研菌科是否通过产生脂多糖或其他途径参与HCC的发生,还需要进一步的研究与探索。
既往研究[30]表明,小鼠结肠胆汁酸水平与厚壁菌门中产生DCA的7α-脱羟基细菌呈正相关,后者指梭菌目细菌,主要包括Ruminococcaceae科和毛螺菌科[31]。本研究发现,与肝硬化组相比,隶属于梭菌目的Christenellaceae科细菌在HCC组肠道内相对丰度明显偏高(P<0.05),而Ruminococcaceae科细菌在HCC组肠道内占比也与肝硬化组不同(肝硬化组10.36% vs HCC组15.51%,P>0.05),或可认为这些细菌通过产生脱氧胆酸导致HCC的发生,但未能发现2组患者胆汁酸水平有显著差异,考虑可能与纳入病例数较少有关。此外,本研究发现HCC组患者肠道内梭菌属所占比例较肝硬化组明显偏低(P<0.05),该差异对肝硬化基础上发生肝细胞癌的影响尚待进一步研究。
Islam等[30]发现小鼠结肠中胆汁酸浓度降低与肠杆菌科等病原菌过生长有关,既往也已明确大肠埃希氏菌(E.coli)增多与肝硬化患者发生HCC有关[22,32]。本研究发现肝硬化组患者肠道内肠杆菌科某属细菌所占比例与HCC组不同(2.52% vs 7.22%,P>0.05),考虑主要原因是E.coli是隶属于埃希氏菌属的大肠杆菌种细菌,而本研究只分析到了属水平的细菌。隶属于变形菌门的其他细菌在肝硬化患者发生HCC的过程中是否起到与E.coli相似的作用,尚待进一步研究。
综上所述,与单纯肝硬化患者相比,肝硬化基础上发生HCC者肠道菌群部分组成有显著差异,但这些差异与肝硬化基础上发生HCC之间相互作用的具体机制尚不明朗。另外,本研究纳入病例数较少,还需要扩大例数、宏基因组学分析等进一步研究探索肠道菌群与肝硬化基础上HCC发生的关系,为HCC的预防探索新的途径。
[1] YE SL. Current challenges to primary liver cancer research[J]. J Clin Hepatol, 2015, 31(6): 819-823. (in Chinese) 叶胜龙. 原发性肝癌研究当前面临的挑战[J]. 临床肝胆病杂志, 2015, 31(6): 819-823.
[2] QIN N, YANG F, LI A, et al. Alterations of the human gut microbiome in liver cirrhosis[J]. Nature, 2014, 513(7516): 59-64.
[3] CHEN Y, YANG F, LU H, et al. Characterization of fecal microbial communities in patients with liver cirrhosis[J]. Hepatology, 2011, 54(2): 562-572.
[4] LU H, WU Z, XU W, et al. Intestinal microbiota was assessed in cirrhotic patients with hepatitis B virus infection. Intestinal microbiota of HBV cirrhotic patients[J]. Microb Ecol, 2011, 61(3): 693-703.
[5] Ministry of Health of the People′s Republic of China. Diagnosis, management, and treatment of hepatocellular carcinoma (V2011)[J]. J Clin Hepatol, 2011, 27(11): 1141-1159. (in Chinese) 中华人民共和国卫生部. 原发性肝癌诊疗规范(2011年版)[J]. 临床肝胆病杂志, 2011, 27(11): 1141-1159.
[6] CAPORASO JG, KUCZYNSKI J, STOMBAUGH J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nat Methods, 2010, 7(5): 335-336.
[7] Greengenes. The greengenes database downloads[DB/OL]. http://greengenes.secondgenome.com/downloads/database/13_5.
[8] TAO X, WANG N, QIN W. Gut Microbiota and Hepatocellular Carcinoma[J]. Gastrointest Tumors, 2015, 2(1): 33-40.
[9] QIN J, LI R, RAES J, et al. A human gut microbial gene catalogue established by metagenomic sequencing[J]. Nature, 2010, 464(7285): 59-65.
[10] MADSEN BS, HAVELUND T, KRAG A. Targeting the gut-liver axis in cirrhosis: antibiotics and non-selective beta-blockers[J]. Adv Ther, 2013, 30(7): 659-670.
[11] SUNG JY, SHAFFER EA, COSTERTON JW. Antibacterial activity of bile salts against common biliary pathogens. Effects of hydrophobicity of the molecule and in the presence of phospholipids[J]. Dig Dis Sci, 1993, 38(11): 2104-2112.
[12] GUNNARSDOTTIR SA, SADIK R, SHEV S, et al. Small intestinal motility disturbances and bacterial overgrowth in patients with liver cirrhosis and portal hypertension[J]. Am J Gastroenterol, 2003, 98(6): 1362-1370.
[13] WIEST R, LAWSON M, GEUKING M. Pathological bacterial translocation in liver cirrhosis[J]. J Hepatol, 2014, 60(1): 197-209.
[14] LI DY, YANG M, EDWARDS S, et al. Nonalcoholic fatty liver disease: for better or worse, blame the gut microbiota?[J]. JPEN J Parenter Enteral Nutr, 2013, 37(6): 787-793.
[15] SEKI E, de MINICIS S, OSTERREICHER CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis[J]. Nat Med, 2007, 13(11): 1324-1332.
[16] LOO TM, KAMACHI F, WATANABE Y, et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity[J]. Cancer Discov, 2017, 7(5): 522-538
[17] BRANDI G, de LORENZO S, CANDELA M, et al. Microbiota, NASH, HCC and the potential role of probiotics[J]. Carcinogenesis, 2017, 38(3): 231-240.
[18] HUANG XF. The role of bile acid signaling in liver carcinogenesis and liver regeneration[D]. Fuzhou: Fujian Med Univ, 2007. (in Chinese) 黄雄飞. 胆酸信号与肝癌发生和肝脏再生[D]. 福州: 福建医科大学, 2007.
[19] YOSHIMOTO S, LOO TM, ATARASHI K, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome[J]. Nature, 2013, 499(7456): 97-101.
[20] YU LX, YAN HX, LIU Q, et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents[J]. Hepatology, 2010, 52(4): 1322-1333.
[21] DAPITO DH, MENCIN A, GWAK GY, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4[J]. Cancer Cell, 2012, 21(4): 504-516.
[22] ZHANG HL, YU LX, YANG W, et al. Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats[J]. J Hepatol, 2012, 57(4): 803-812.
[23] MITSUOKA T. Bifidobacteria and their role in human health[J]. J Ind Microbiol, 1990, 6(4): 263-267.
[24] SHETH AA, GARCIA-TSAO G. Probiotics and liver disease[J]. J Clin Gastroenterol, 2008, 42 (Suppl 2): s80-s84.
[25] MALAGUARNERA M, GRECO F, BARONE G, et al. Bifidobacterium longum with fructo-oligosaccharide (FOS) treatment in minimal hepatic encephalopathy: a randomized, double-blind, placebo-controlled study[J]. Dig Dis Sci, 2007, 52(11): 3259-3265.
[26] ZHANG M, ZHENG M, WU Z, et al. Alteration of gut microbial community after N,N-Dimethylformamide exposure[J]. J Toxicol Sci, 2017, 42(2): 241-250.
[27] HE J, LIU J, KONG Y, et al. Serum activities of liver enzymes in workers exposed to sub-TLV levels of dimethylformamide[J]. Int J Occup Med Environ Health, 2015, 28(2): 395-398.
[28] KIM KW, CHUNG YH. Hepatotoxicity in rats treated with dimethylformamide or toluene or both[J]. Toxicol Res, 2013, 29(3): 187-193.
[29] LIU HX, ROCHA CS, DANDEKAR S, et al. Functional analysis of the relationship between intestinal microbiota and the expression of hepatic genes and pathways during the course of liver regeneration[J]. J Hepatol, 2016, 64(3): 641-650.
[30] ISLAM KB, FUKIYA S, HAGIO M, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats[J]. Gastroenterology, 2011, 141(5): 1773-1781.
[31] RIDLON JM, ALVES JM, HYLEMON PB, et al. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship[J]. Gut Microbes, 2013, 4(5): 382-387.
[32] GRAT M, WRONKA KM, KRASNODEBSKI M, et al. Profile of gut microbiota associated with the presence of hepatocellular cancer in patients with liver cirrhosis[J]. Transplant Proc, 2016, 48(5): 1687-1691.
引证本文:ZHOU LX, HAN T, LIU BW, et al. A preliminary analysis of composition and structure of intestinal microbiota in patients with liver cirrhosis or hepatocellular carcinoma[J]. J Clin Hepatol, 2017, 33(9): 1740-1744. (in Chinese) 周冷潇, 韩涛, 刘保文, 等. 肝硬化及肝细胞癌患者肠道微生态组成和结构的初步分析[J]. 临床肝胆病杂志, 2017, 33(9): 1740-1744.
(本文编辑:王 莹)
Apreliminaryanalysisofcompositionandstructureofintestinalmicrobiotainpatientswithlivercirrhosisorhepatocellularcarcinoma
ZHOULengxiao,HANTao,LIUBaowen,etal.
(TheThirdCentralClinicalCollegeofTianjinMedicalUniversity,Tianjin300170,China)
ObjectiveTo investigate the differences in intestinal microbiota between patients with liver cirrhosis and those with hepatocellular carcinoma (HCC) complicated by liver cirrhosis, as well as the association between the change in intestinal microbiota and the development of HCC.MethodsA total of 35 patients with chronic liver diseases who were hospitalized in Department of Hepatology in Tianjin Third Central Hospital from December 2015 to May 2016 were enrolled, among whom 20 patients had liver cirrhosis (liver cirrhosis group) and 15 had HCC complicated by liver cirrhosis (HCC group). Fecal samples were collected from all patients, total bacterial DNA was extracted, and Roche 454 sequencing was used to determine the sequence of the V3-V6 viable regions of 16S rDNA. A bioinformatics analysis was also performed (species taxonomy, abundance analysis, and diversity analysis). Thet-test was used for comparison of continuous data between groups, and the Mann-WhitneyUtest was used for comparison of categorical data between groups.ResultsThe mean number of operational taxonomic units (OTUs) in the samples from the 20 liver cirrhosis patients was 306.50±163.76, and that in the samples from the 15 HCC patients was 357.24±168.85; there were no significant differences in the number of OTUs and alpha diversity between the two groups of patients (bothP>0.05). The bacteria in fecal samples included Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria. Genus-species and composition analyses showed that there was a significant difference in relative abundance of various bacteria in the intestine between the liver cirrhosis group and the HCC group, and compared with the HCC group, the liver cirrhosis group had significant increases in the proportions of Actinobacteria (0.21% vs 0.06%,U=89.000,P=0.043), Bifidobacterium (0.16% vs 0.04%,U=90.000,P=0.046), and Clostridium (0.13% vs 0.08%,U=90.000,P=0.046) and significant reductions in the proportions of Rikenellaceae (0.58% vs 2.30%,U=82.000,P=0.023) and Christenellaceae (0.01% vs 0.08%,U=84.500,P=0.028).ConclusionThere are significant differences in the composition of intestinal microbiota between patients with liver cirrhosis and those with HCC complicated by liver cirrhosis, while the specific mech anisms of the interaction between the differences and HCC with liver cirrhosis remained unclear.Keywords:liver cirrhosis; carcinoma, hepatocellular; enterobacteriaceae
10.3969/j.issn.1001-5256.2017.09.022
2017-03-20;
2017-05-02。
国家科技重大专项(2016ZX10002008-07);天津市科技计划项目(13RCGFSY19200)
周冷潇(1991-),女,主要从事肝脏疾病的相关研究。
韩涛,电子信箱:hantaomd@126.com。
R575.2; R735.7
:A
:1001-5256(2017)09-1740-05
