呋喃与(HF)n (n=1-3)分子间氢键相互作用研究
2017-09-06吴思展张福星
吴思展,张福星
(1.铜仁学院 材料与化学工程学院,贵州 铜仁 554300;2.铜仁学院 农林工程与规划学院,贵州 铜仁 554300)
呋喃与(HF)n (n=1-3)分子间氢键相互作用研究
吴思展1,张福星2
(1.铜仁学院 材料与化学工程学院,贵州 铜仁 554300;2.铜仁学院 农林工程与规划学院,贵州 铜仁 554300)
采用MP2/6-311++G(d, P)方法对单体HF、呋喃(C4H4O),以及它们形成的复合物C4H4O与(HF)n(n= 1-3)的结构和相互能量等进行了研究。结果显示复合物C4H4O与(HF)n(n=1-3)中随着HF分子的增多,复合物结合能增大,但C4H4O与HF形成的单个氢键键能减小,且当n=3时C4H4O已不能同时与3个HF形成三个分子间氢键。自然轨道(NBO)显示,C4H4O与(HF)n(n=1-2)中随着n值的增大,C4H4O中的O原子与FH形成的单个氢键相互作用显著减小。C4H4O与(HF)n(n=1-2)中形成的氢键具有一定的方向性和饱和性。
HF;C4H4O;氢键;自然键轨道
氢键概念首先是由Hvggins在1920年率先提出的,随后Latiwerhe和Rodebush将氢键概念成功应用于解释水沸点的反常现象。1939年,随着Pauling编著的《化学键的本质》一书的出版,使氢键被化学界广泛接受[1-2]。近年来由于氢键等分子间弱相互作用在物质分子,以及生命大分子的结构、性质和功能等方面起着至关重要的作用,使其在生物、化学、物理等领域受到广泛关注。近年来的研究文献报道,科研工作者们利用氢键的特性,将氢键应用于生物分子识别、分子及晶体工程、分子簇的形成、化学反应过程、材料设计和分子自组装等[3-6]。但是关于分子间氢键的饱和性和方向性的研究较少,对于氢键的饱和性和方向性问题科学界总说纷纭。为了研究这个问题,我们采用从头算方法对C4H4O与( HF )n(n = 1-3)复合物的氢键相互作用进行了研究。
1 计算方法
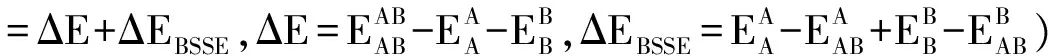
2 结果与讨论
2.1 复合物构型
为了考察呋喃上O与HF分子形成的氢键是否具有饱和性和方向性,在优化前根据C4H4O与(HF)n(n=1-3)复合物的分子数构造了三种复合物的输入构型,如图1所示。图A是C4H4O/( HF )复合物的构型,此时1个HF分子以0°角正对呋喃分子中的O原子,复合物为一字形构型。图B是C4H4O/(HF)2复合物的构型,此时2个HF分子与呋喃分子中的O原子呈90°相对,复合物程T字形构型。图C是C4H4O/( HF)2复合物的构型,此时3个HF分子与呋喃分子中的O原子垂直相对,复合物程十字形构型。复合构型经过MP2/6-311++g**方法优化后,复合物的稳定构型如图2所示。图D、E和F的结果分别对应图A、B和C的构型。C4H4O/( HF )经过优化后构型总体结构没有发生明显变化,至是呋喃上O原子与HF分子中H的距离缩小了,HF中F-H键的键长由原来的0.09166 nm伸到0.09264 nm,这是由于形成了氢键,F-H键发生了红移现象。C4H4O/(HF)2复合物经过优化后构型发生了较小的变化,HF与呋喃分子形成的二面角由原先的90°变为125°,HF中F-H键的键长由原来的0.09166nm拉伸到0.09226nm,这也是由于形成了氢键,F-H键发生了红移现象,其红移程度没有前面二具体的氢键明显。由图2中的F图可以看出,经过优化后,只有一个HF分子正对呋喃的O原子,其余两个HF分子由于受到其它原子的排斥力的作用都不能与O原子形成氢键。这初步说明氢键具有一定的方向性和饱和性。
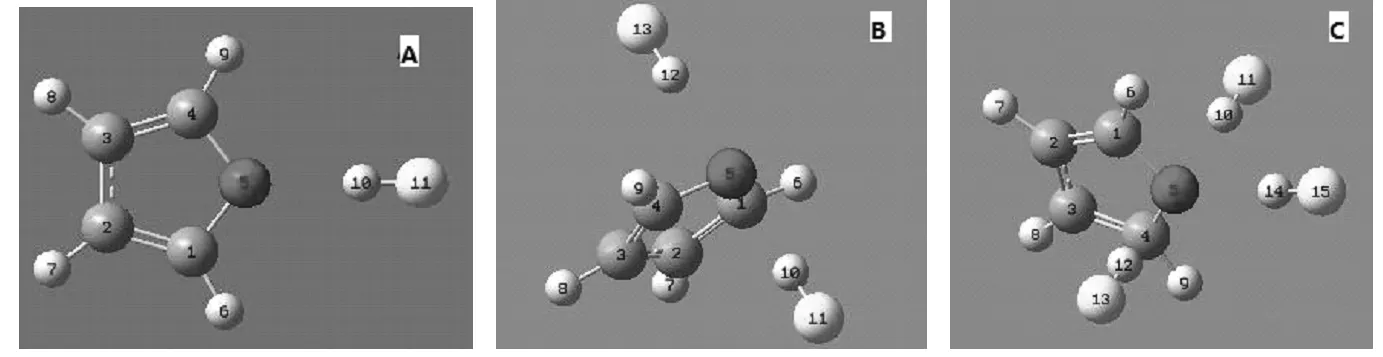
图1 C4H4O与(HF)n(n=1-3)复合物优化前的几何结构
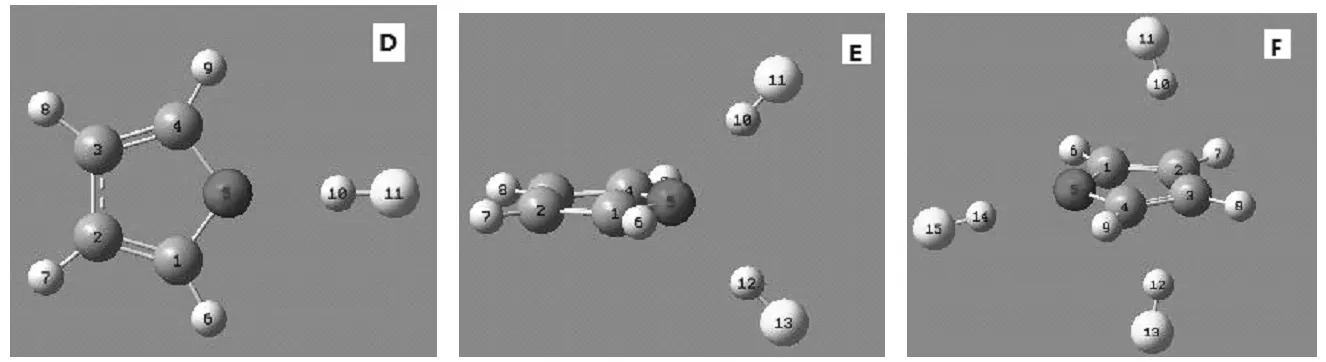
图2 C4H4O与( HF )n (n=1-3)复合物优化后的几何结构
2.2 复合物结合能

表1 C4H4O/(HF)n (n = 1-2)复合物的氢键结合能
表1为C4H4O/(HF)n(n=1-2)复合物的氢键结合能,ΔE为复合物的氢键稳定化能,ΔE'是经过基组重叠误差(BSSE)校正后的结合能。由表1可知,基组重叠误差对氢键这类弱相互作用的影响比较显著,计算此类弱相互作用需做BSSE校正。从表中可知C4H4O/(HF)的氢键结合能比C4H4O/(HF)2的小,但是C4H4O/(HF)2复合物有两个氢键,平均计算每个氢键的键能,前者的氢键的能量大于后者的能量。C4H4O/(HF)2复合物一个氢键的能量为13.9034 KJ/mol其显著低于C4H4O/(HF)的21.0499 KJ/mol,这说明呋喃上的O原子随着与HF分子形成氢键,其氢键的强度显著减弱。
2.3 复合物的自然键轨道(NBO)分析
为了分析C4H4O/(HF)n(n = 1-2)复合物的氢键相互作用本质,通过计算C4H4O/(HF)n(n = 1-2)体系的自然轨道(NBO),表2给出了C4H4O/(HF)和C4H4O/(HF)2复合物中氢键的电子受体轨道j和电子供体轨道i相互作用的二阶稳定化能E(2)。E(2)越大表明i轨道提供电子给j轨道的程度越大,氢键的相互作用越大。表2中E(2)的稳定化能的大小与前面的氢键结合能相似呋喃中的O原子与HF分子形成氢键的数目增加,其氢键能和稳定化能也急剧下降。这说明氢键具有一定的方向性和饱和性。
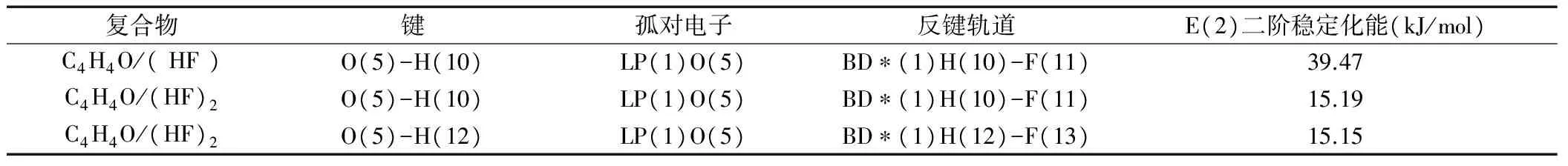
表2 C4H4O/(HF)n (n = 1-2)复合物的二阶稳定化能E(2)
3 结论
通过采用MP2/6-311++G(d, P)方法对单体HF、呋喃(C4H4O),以及它们形成的复合物C4H4O与(HF)n(n = 1-3)的结构、氢键相互能量和NBO分析等进行了研究,结果表明随着呋喃O原子与HF分子形成氢键的数目增加,造成每个氢键的键能降低。当HF分子超过三个以上(含三个),呋喃O原子只与一个分子形成氢键,这表明氢键具有一定的饱和性和方向性。
[1] 瓦塞 J,特鲁布拉德 K N,诺布勒著 C M. 大一化学[M]. 北京化工学院应用化学系无机化学教研室, 译. 北京: 科学出版社,1987.
[2] Jeffrey G A, Saenger W. Hydrogen Bonding in Biological Structures[M]. New York: Springer-verlag, 1991.1-5, 8-9.
[3] 王海燕,曾艳丽,孟令鹏,等. 有关氢键理论研究的现状及前景[J]. 河北师范大学学报:自然科学版,2005,29(2):177-181.
[4] 伍宏伟,陈亚运,饶才辉,等. 含CH基的阴离子受体[J].化学进展,2016,28(10):1501-1514.
[5] 杜本妮,张为明. FOCl与(H2O)n(n=1-4)分子间相互作用的理论研究[J]. 江苏师范大学学报:自然科学版,2012,30(2):53-59.
(本文文献格式:吴思展,张福星.呋喃与(HF)n(n=1-3)分子间氢键相互作用研究[J].山东化工,2017,46(10):7-8.)
Study on the Molecular Hydrogen BondingInteraction Between C4H4O and(HF)n(n=1-3)
WuSizhan1,ZhangFuxing2
(1. School of Material and Chemical Engineering, Tongren University, Tongren 554300, China;2. School of Agriculture and Forest Engineering and Planning, Tongren University, Tongren 554300, China)
The hydrogen bond energy, geometrical structure and NBO of complexes were studied using MP2/6-311++G(d, P) method. The result showed that the energy of the complexes increased with the increasing HF molecules. But the single hydrogen bond energy in the complexes reduced with the increasing HF molecules. The NBO analysis of the complex showed the same result as the hydrogen energy. This study revealed that the hydrogen bonds had orientation and saturation in the C4H4O/(HF)ncomplexes.
HF;C4H4O; hydrogen bond;NBO
2017-03-07
铜仁学院校级科研项目"氢键质子受体饱和性量子化学研究"(TS1015)
吴思展(1980—),广东揭西县人,铜仁学院材料与化学工程学院副教授,主要从事理论计算、催化剂和纳米材料等先进材料的制备和应用研究。
O641.1
A
1008-021X(2017)10-0007-02
