固相萃取-高效液相色谱法测定婴幼儿乳粉中维生素D3的含量
2017-09-04刘家阳
黄 旭,袁 波,刘家阳
(1. 沈阳药科大学,辽宁 沈阳 110032;2.辽宁省食品检验检测院,辽宁 沈阳 110015)
固相萃取-高效液相色谱法测定婴幼儿乳粉中维生素D3的含量
黄 旭1,2,袁 波1,刘家阳2
(1. 沈阳药科大学,辽宁 沈阳 110032;2.辽宁省食品检验检测院,辽宁 沈阳 110015)
建立一种使用高效液相色谱紫外检测器测定婴幼儿乳粉中维生素D3含量的方法。样品经高温皂化反应后,将皂化液通过 固相萃取柱,对目皂化液中的维生素D3进行提取,提取液经浓缩后,再经过正相硅胶柱净化,氮吹复溶,通过Zorbax C18色谱柱分离,以外标法定量。维生素D3标准溶液,在曲线范围内呈良好的线性关系,相关系数r2均大于0.999;样品3水平加标回收率分别为97%~102%,6次重复测定RSD小于2.0%;检出限为1.0μg /100g,定量限为3.0μg /100g。
固相萃取;婴幼儿乳粉;维生素D3;正相净化
维生素D是脂溶性维生素的一类[1],自然界中广泛存在,其最重要的两种形态是维生素D2(麦角钙化醇)和维生素D3(胆钙化醇)。而维生素D3的活性最高,婴幼儿乳粉中所添加的维生素D均是以维生素D3的形式存在,本文所讨论的维生素D也均指维生素D3。维生素D3对人体十分重要,尤其对婴幼儿来说就更加不可或缺,如果婴幼儿缺少维生素D3,那么容易造成婴幼儿缺钙、佝偻病等疾病[2];而如果婴幼儿维生素D3摄入过多,又会造成婴幼儿的头疼、心律不齐等症状,因此合理的维生素D3的摄取,对于婴幼儿来说就极为重要。
现行有效的国家标准[3]中对维生素D3检测规定的方法是基于皂化法,对皂化液进行液液萃取,合并分次的萃取液后经旋转蒸发装置浓缩后定容,再使用正相色谱,对浓缩液中的干扰物质进行正相分离除杂,最终上机液进入液相色谱分析。
上述方法操作过程复杂,液液萃取容易出现待测物提取不完全,两相液面出现乳化等现象;而正相色谱净化往往需要较专业的半制备液相色谱,以上两点均不利于大批量,多批次检验操作。本文讨论了使用固相萃取代替液液萃取的前处理,且提取液的净化使用正相硅胶固相萃取柱代替正相色谱的净化方法,大大降低了检验的繁琐程度,提高了检验效率,适用于实验室大批量,多批次检验的要求。
1 实验部分
1.1 仪器与试剂
美国Waters E2695高效液相色谱,包括四元梯度泵,VWD检测器,固相萃取装置(Agilent),分析天平(Mettler),Milli-Q超纯水(美国Millipore公司);旋转混合仪(IKEA),恒温振荡水浴(FOSS)、旋转蒸发仪(布奇)。
维生素D3标准品(中国食品药品检定研究院,201208),EXtrelut NT20 固相萃取柱(默克公司)、Waters Silica硅胶固相萃取柱,甲醇、正己烷、乙酸乙酯、乙醇、乙腈为色谱纯(Fisher公司),氢氧化钾、BHT(2,6-二叔丁基-4-甲基苯酚)、抗坏血酸等为分析纯(国药集团化学试剂有限公司)。
1.2 色谱条件
Agilent Zorbax C18色谱柱(4.6mm×250mm,粒径5μm),流动相为甲醇,流速为1.0mL/min,进样量100μL,检测波长为:264nm,色谱柱温度为常温。
1.3 标准溶液配置
标准品储备液:准确称取维生素D3标准品10.0mg,用色谱纯无水乙醇溶解并定容至100mL,使其浓度约为100μg/mL,转移至100mL的棕色试剂瓶中,临用前用紫外分光光度法校正其浓度。
标准品中间液:准确吸取维生素D3标准储备溶液10.00mL,用甲醇稀释并定容至100mL的棕色容量瓶中,浓度为10.0μg/mL。
标准品标准使用液:分别准确吸取维生素D3标准中间使用液0.50,1.00,2.00,5.00,10.00mL于100mL 棕色容量瓶中,用甲醇定容至刻度混匀。此标准系列工作液浓度分别为0.05,0.10,0.20,0.50,1.00μg/mL。
1.4 样品的前处理过程
1.4.1 皂化过程
1.4.1.1 不含淀粉的样品
准确称取均质后的样品10g(精确至0.01g)于250mL棕色碘量瓶中,加入约20mL水,加入1g抗坏血酸,加入50mL无水乙醇,加入5g氢氧化钾,将上述溶液摇晃均匀,慢慢向瓶底部冲入氮气,将瓶口盖紧密封,放恒温振荡水浴中,85℃边振荡边进行皂化反应30min。反应完全后,将溶液放置至室温,用水将全部皂化液转移到100mL容量瓶中,用水定容至刻度,摇均。
1.4.1.2 含淀粉的样品
称取均质后的样品10g(精确至0.01g) 于250mL棕色碘量瓶中,加入20mL温水混匀,加入0.5g淀粉酶,放入60℃水浴避光恒温振荡30min后,然后按照1.4.1.1的步骤依次进行样品皂化。
1.4.2 固相萃取过程
将固相萃取柱放置在固定的试管架上,将下口敞开,准确移取20.0mL皂化液至固相萃取柱上,待皂化液慢慢留下,待溶液完全被吸附,但不能流出固相萃取柱,等待10min后,使用含有0.1%BHT(质量分数)的正己烷进行洗脱,洗脱过程中,固相萃取柱下方应链接注射器针头,并将针头下方放入150mL棕色旋蒸瓶中,分4次向固相萃取柱上添加正己烷,每次25mL,将所有洗脱液全部收集。
1.4.3 浓缩过程
将旋蒸瓶放置在旋转蒸发上,水浴温度调至40℃,进行旋转蒸发,待瓶内液体近干时,使用少量正己烷将瓶内液体全部转移至10mL离心管中备用。
1.4.4 净化过程
取硅胶固相萃取柱,使用5mL正己烷活化,抽干后,将1.4.3制得浓缩液全部转移至固相萃取柱上,过柱抽干后,用乙酸乙酯-正己烷(5+95)溶液5mL淋洗杂质并弃去,使用乙酸乙酯-正己烷(30+70)溶液5mL洗脱,收集全部洗脱液于离心管中,40℃水浴氮气吹干,使用1.0mL甲醇复溶,旋涡30s。将此溶液过0.45μm滤膜上机测定,得到如下色谱图1。
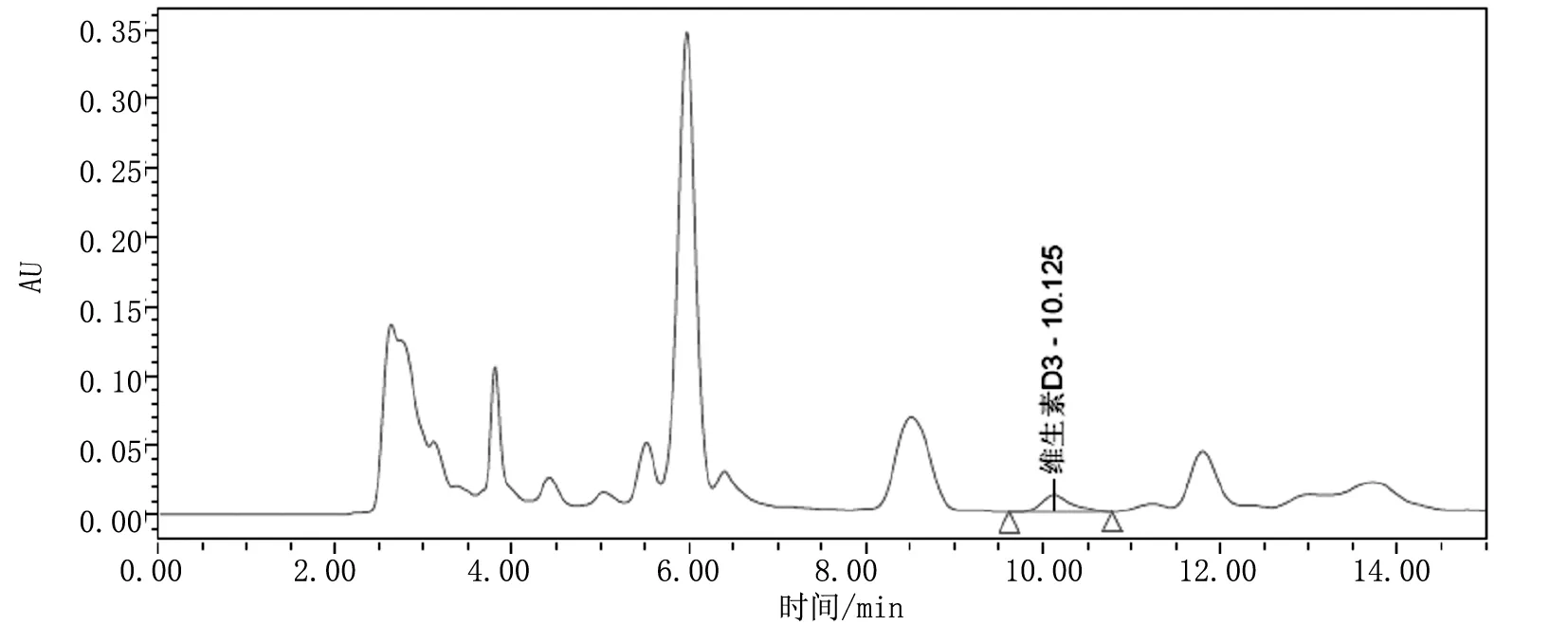
图1 样品处理液上机色谱图
2 结果及讨论
2.1 前处理过程
2.1.1 待测物的提取
2.1.1.1 脂肪类物质的清除
社会团体推动型联盟定位为学会协会等社会团体组织在产业层面落实国家创新体系的途径和举措,如山西、重庆、广西、北京、内蒙古、武汉、香港地区的BIM联盟由各省建筑领域相关协会主导发起,推动本省联盟成立及开展相关工作,联盟承担着一定政府职能。
婴幼儿乳粉中有很多脂肪类物质,而维生素D3作为脂溶性维生素和这些脂肪组分结合紧密,为了能将维生素D3提取出来,就需要将样品中的脂肪类物质除去,皂化就是在强碱性和高温的条件下,将脂肪类物质转化为醇类和盐,本方法采用皂化法除去脂肪。
2.1.1.2 皂化前充入氮气、加入抗坏血酸和乙醇的作用
维生素D3容易被氧化,在皂化前应使用氮气排空皂化瓶中的空气。皂化体系中加入抗氧化剂-抗坏血酸,目的是防止维生素D3在皂化过程中被溶液体系中溶解的氧气氧化。乙醇在皂化反应过程中的作用有两个,第一作为皂化反应的催化剂,作为载体可以使氢氧化钠溶液和油脂充分结合以达到加快反应的目的,第二增加待测物在溶液体系中的溶解性,保证了维生素D3的充分提取。
2.1.2 固相萃取过程
2.1.2.1 固相萃取的原理
EXtrelut NT20 固相萃取柱其填料为特殊处理的大孔硅藻土,具有很大的孔体积,且具有化学惰性,皂化液放入时,会被这些多孔吸附,当使用弱极性的溶液对固相萃取柱上残留的物质进行洗脱时,只能将极性相对较弱的物质有选择的洗脱下来,而水和无机盐等杂质保留在柱体上。皂化液上样量为20.0mL时,使用100mL的正己烷可将目标待测物完全洗脱下来。
2.1.2.2 固相萃取过程对待测物的保护
维生素D3易于被氧化,为了保证萃取过程维生素D3的不损失,在洗脱液正己烷中加入BHT,BHT是一种良好的抗氧化剂[4],可很好的保护维生素D3,不会在提取过程之中被氧化。
2.1.3 净化
2.1.3.1 传统的净化方式
在现有的方法中,均采用正相色谱使用硅胶色谱柱进行分离,除杂后,将待测组分保留时间的流出成分收集,氮吹复溶后再通过反相色谱测定。传统方法操作复杂,且需要较为专业的半制备液相色谱,不利于日常大批量多批次的前处理操作。
2.1.3.2 正相固相萃取[5]
使用具有极性的正相硅胶固相萃取柱,将含有维生素D3的弱极性溶剂正己烷上样,当通过固相萃取柱时,待测物维生素D3保留在固相萃取柱上,此时维生素D3在体系中属于极性稍强的物质,使用弱极性的淋洗液进行淋洗后,对维生素D3造成干扰的组分被洗出固相萃取柱,再使用极性稍强的洗脱液进行洗脱,可将维生素D3从硅胶柱上洗脱下来,通过氮吹浓缩、复溶后,进入色谱进行分析。在选择淋洗液和洗脱液时,分别尝试了乙酸乙酯、乙醚、丙酮等常用试剂,从试剂友好的角度和洗脱液的浓缩过程来看,最后选定乙酸乙酯和正己烷的混合溶液作为最终洗脱液,可保证对待测组分的良好洗脱。
2.2 液相色谱条件
2.2.1 色谱条件
由于乳粉中成分复杂,为了将目标峰和干扰组分分离的更好,实验选择长度为250 mm的安捷伦ZORBAX C18色谱柱(4.6mm×250mm,粒径5μm),而标准工作液和上机液的浓度较低,因此进样量需要较大,选择进样量为100μL,而二极管阵列检测器灵敏度较低,因此选择紫外检测器进行测定,以甲醇为流动相的条件下,柱温为室温,流速为1.0mL/min,扫描波长设为264nm,进入高效液相色谱分析。
2.2.2 线性方程、方法灵敏度
2.2.2.1 方法的线性关系和检出限
以甲醇作为稀释液,准确移取不同体积的混合标准溶液分别配制成混合标准溶液,标准溶液浓度依次为维生素D3:0.05,0.10,0.20,0.40,1.00μg/mL。
结果表明,维生素D3在范围内有良好的线性关系,线性方程为Y = 3.09×105X + 3.80×103线性系数为0.999。分别以信噪比S/N=3的标准溶液浓度和S/N=10的标准溶液浓度作为样品处理上机液的检出限浓度和定量限浓度,得到维生素D3的检出限和定量限分别为1.0μg/100g、3.0μg/100g。
2.2.2.2 样品平行测定结果精密度选择实验室检毕样品作为样本测定维生素D3的含量,平行测定六次,得到结果分别为维生素D:9.50,9.29,9.32,9.63,9.17,9.30μg/100g,相对标准偏差(RSD)为1.91%。
2.2.2.3 回收率
向已知浓度的婴幼儿乳粉样品中在高、中、低3个浓度添加水平下进行回收试验,平行3份。标准物质添加量,回收率平均值见表1。

表1 已知浓度样品三水平加标回收率
2.2.2.4 实际样品的测试分析结果
选择实验室日常试验剩余样品,按照上述方法进行维生素D的测定,得到结果见表2。
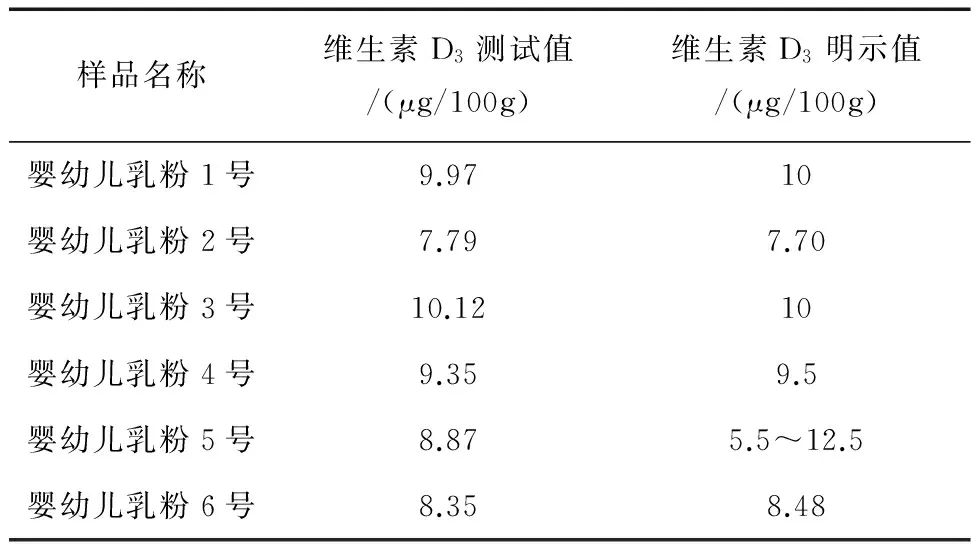
表2 实验室检测样品标示值和实际测定值
3 小节
婴幼儿乳粉中脂溶性维生素的检验是一个难题,不同实验室之间的检测结果也往往出现很大偏差,究其原因就是因为国标中规定的检验方法可操作性差,检测周期较长,不能满足大批量多批次工作任务的需求。本方法的建立,简化了操作步骤,提高了实验效率,且经过方法学验证,能够满足日常检验,在实际样品测定中,检测结果也能在样品的明示值范围之内,可以作为实验室提检验婴幼儿乳粉中维生素D3检验方法使用。
[1] 尉全平. 维生素D的生物学作用[J]. 河北医药,2013 (06):912-914.
[2] 胡冠琼,池美珠,孙彩霞. 孕母维生素D缺乏对婴幼儿生长发育的影响[J].中国妇幼保健,2015 (15):2350-2352.
[3] 中华人民共和国卫生部.GB 5413.9-2010 婴幼儿食品和乳品中维生素A、D、E的测定[S].北京: 中国标准出版社,2010.
[4] 汤务霞,陈明涛,阙晓莉. 抗氧化剂BHT和维生素E对菜籽油的抗氧化研究[J]. 中国食品添加剂,2011 (04):59-62.
[5] 陆慧媛,姜 珊,张 烈,等. 商品化正相硅胶固相萃取柱-气相色谱法检测橄榄油中的蜡含量[J]. 食品安全质量检测学报,2016 (11):4496-4503.
(本文文献格式:黄 旭,袁 波,刘家阳.固相萃取-高效液相色谱法测定婴幼儿乳粉中维生素D3的含量[J].山东化工,2017,46(08):82-84.)
Determin vitamin D3in Infant milk powder with SPE-HPLC
HuangXu1,2,YuanBo1,LiuJiayang2
(1. Shenyang Pharmaceutical University, Shenyang 110032,China;2.Liaoning Institute For Food Control, Shenyang 110015,China)
Establish a method for detection of vitamin D3in Infant milk powder with HPLC-VWD。After saponification, pipette the saponified solution to the SPE cartridge, elute and collect the solution and evaporate the solution, and after purification by normal phase silica gel column, redissolution and inject into the HPLC to analyse, separated by Zorbax C18 column and calculate by the external standard method. The solution had good linear relationship within linear ranges, the correlation coefficients of the standard curve were 0. 999, the recoveries at three levels were from 97%~102%, and the relative standard deviations ( RSD ) were less than 2.0%; the LOD is 1.0μg /100g and the LOQ is 3.0μg /100g.
solid phase extraction; infant milk powder;vitamin D3;positive phase purification
2017-03-03
黄 旭(1982—),辽宁沈阳人,工程师,主要从事食品检验和食品检测方法开发的工作。
O657.7+2
A
1008-021X(2017)08-0082-03
