2-甲基-1-硝基萘激发态分子内氢转移的瞬态吸收光谱及其理论计算研究
2017-09-03关怀昱薛佳丹
关怀昱,薛佳丹
(浙江理工大学理学院,杭州 310018)
2-甲基-1-硝基萘激发态分子内氢转移的瞬态吸收光谱及其理论计算研究
关怀昱,薛佳丹
(浙江理工大学理学院,杭州 310018)
利用纳秒瞬态吸收光谱技术和密度泛函理论(DFT)、完全活化空间自洽场(CASSCF)等计算方法,研究了2-甲基-1-硝基萘(2M1NN)在有机溶剂中的激发态分子内氢转移反应。通过实验获得了2M1NN的瞬态吸收光谱和动力学常数,400 nm处拟合得到3个衰减时间常数,其中25 μs的物种指认为三重态分子内氢转移后生成的顺式硝酸亚基中间体。实验获得二茂铁猝灭三重态2M1NN的速率常数为1.77×1010mol-1·dm3·s-1。理论计算预测了可能的氢转移通道,推测分子内氢转移的机理为三重态2M1NN发生分子内氢转移生成双自由基中间体,再系间窜越生成硝酸亚基中间体。
硝基多环芳烃;三重态;纳秒瞬态吸收;CASSCF;激发态分子内氢转移
0 引 言
硝基多环芳烃(nitro-polycyclic aromatic hydrocarbons, NPAHs)属于持久性有机污染物,自然界中NPAHs的结构有两百多种。其形成机理尚未完全明确,一般认为主要来自于汽油或柴油燃料的不完全燃烧[1-2]。NPAHs能溶于气溶胶的小液滴中,也可以被吸附在固相微粒表面,人体吸入后将导致染色体异常,增加癌症的发病率[3-4]。光化学反应是NPAHs在自然界中的主要降解途径[5],因此NPAHs光化学反应研究是重要的课题。激发单重态的NPAHs一般有两条衰减路径:其一,发生超快系间窜越到达三重态;其二,发生光解离反应生成芳氧自由基,进而反应生成稳定的产物[5-6]。NPAHs系间窜越大约发生在200 fs左右,这是由于S1(π,π*)和Tn(n,π*)之间能隙小,并且有强的自旋-轨道耦合。
研究表明T1态硝基苯及其衍生物电子跃迁类型为3(n,π*)[7],而T1态NPAHs为3(π,π*)类型[5,8]。邻位基团含氢的硝基苯衍生物T1(n,π*)具有分子内氢转移性质,如邻硝基甲苯[9]、邻硝基苯甲醛[10]等。利用这一性质,这类分子可作为光致离去保护基团(photoremovable protecting groups, PPGs),并且广泛应用于生物分子捕获、DNA及其他生物分子合成等方面[11-12]。以邻硝基甲苯为例,T1(n,π*)的分子内氢转移机理为:硝基直接夺取邻位甲基上的氢原子形成双自由基中间体,该步反应的时间常数约为430 ps;然后中间体发生系间窜越形成基态产物,这一步的反应时间常数约为1.3 ns[12]。
目前对于NPAHs的激发态分子内氢转移反应还未见报道。本文利用纳秒瞬态吸收实验,以及含时密度泛函理论(TD-DFT)、完全活化空间自洽场理论(CASSCF)等计算方法,研究了2-甲基-1-硝基萘(2M1NN)发生激发态分子内氢转移的机理。
1 实验部分
1.1 主要实验试剂及仪器
2-甲基-1-硝基萘(99%,Sigma-Aldrich公司),二茂铁(99%,上海百灵威公司),乙腈(99.9%,Spectrum公司)。
Cary 50型紫外可见吸收光谱仪(美国Varian公司);LP-920型瞬态吸收光谱仪(英国Edinburgh仪器公司);Lab-150-10/Nd:YAG型激光器(美国Spectra-Physics公司)产生的354.7、266.0 nm激光(脉宽10 ns)提供激发光,450 W氙弧灯提供探测光,两束光垂直会聚在1 cm×1 cm的石英比色皿样品上。瞬态物种光信号经Czerny-Turner闪耀光栅单色仪分光,由光电倍增管(Hamamatsu R928型)或增强电荷耦合装置(ICCD,AndorDH720型)检测,最终传输到电脑终端。瞬态物种的吸光度由朗伯比尔定律得(A(λ,t) =ε(λ)c(t)l=lg(I100/It)。其中ΔA(λ,t)是与波长、时间相关的瞬态物种吸光度;ε(λ)为瞬态物种摩尔消光系数;c(t)为瞬态物种随时间变化的浓度;l为有效吸光光程;I100为激发前探测光透过样品池的光强度;It为激光激发后探测光透过样品池的光强度。由于ΔA是与波长、时间相关的物理量,故瞬态吸收光谱仪有全波段频谱和时域动力学两种工作模式。
瞬态吸收实验中,2M1NN溶液浓度为使激发波长处吸光度达到0.5。溶液在比色皿中通入氩气20 min,以除去溶解的氧气。实验所得数据用Igor或Origin软件处理和拟合。
1.2 理论计算方法
主要采用含时密度泛函理论(TD-DFT)计算物种的垂直跃迁能和振子强度,完全活化空间自洽场方法(CASSCF)计算激发态、过渡态的结构,利用二阶多参考微扰理论(CASPT2)优化其能量,使用Gaussian 09[13]和Molpro[14]软件,在国家超级计算深圳中心的计算机系统进行运算。
2 结果与讨论
2.1 瞬态吸收及动力学实验
图1为2M1NN在除氧的乙腈中被355 nm激光激发后的瞬态吸收光谱。0 ns的光谱中有两个较宽的吸收带,其中心值在420 nm和590 nm,这两个宽吸收带寿命较短,完全衰减后,剩余325、349、382、404 nm处4个比较尖的吸收峰。图1中的插图为0 ns和2 μs的差减光谱,即短寿命物种的光谱,根据文献[8],其由T1态2M1NN(以32M1NN表示,下同)产生。
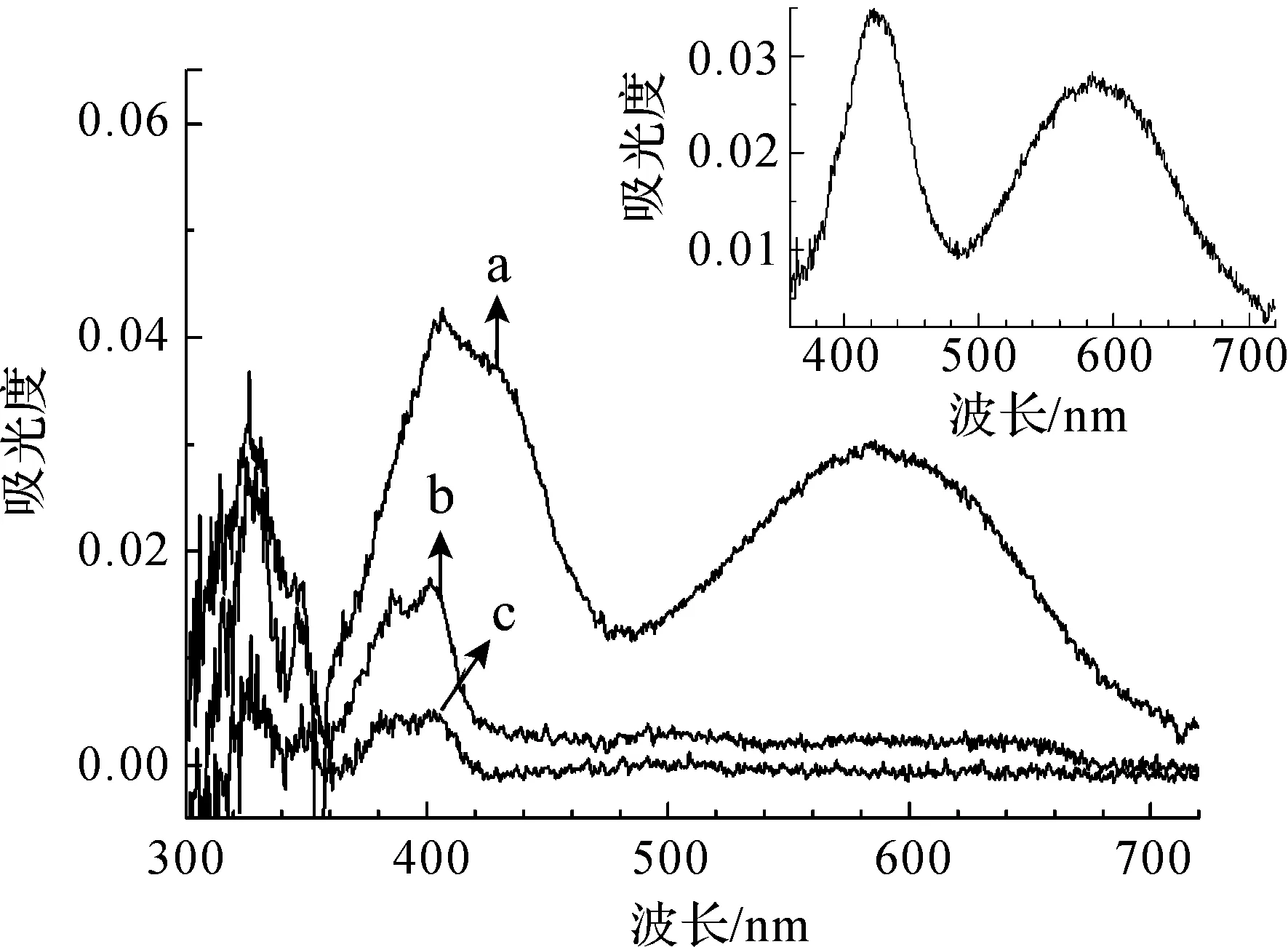
a.时间延迟为0 ns;b.时间延迟为2 μs;c.时间延迟为50 μs图1 2M1NN在乙腈(Ar气氛)中355 nm激发的瞬态吸收光谱注:右上角图为0 ns与2 μs的差谱。
400 nm附近的动力学曲线需要三指数函数拟合,如图2所示,在除氧的乙腈中短寿命物种32M1NN的衰减时间常数约为800 ns,两个长寿命物种的衰减时间常数分别约为25 μs和170 μs。170 μs长寿命物种是2-甲基-1-萘氧自由基[15-16],特征吸收峰位置在384 nm和403 nm。32M1NN和三重态1-硝基萘(以31NN表示,下同)结构相似,但是相同条件下32M1NN的衰减比31NN快得多,时间常数仅是31NN的0.12~0.24倍[17],因此有理由相信32M1NN发生了更快速的反应,而25 μs物种与该反应有关。通过差减光谱获得25 μs物种在400 nm处具有特征吸收。
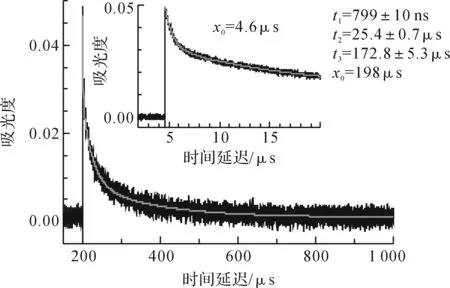
图2 2M1NN在乙腈(Ar气氛)中400 nm吸光度随时间变化的动力学曲线注:右上角图为初始时间延迟的动力学曲线。
32M1NN能够通过能量转移反应被二茂铁猝灭,图3为32M1NN衰减速率常数与二茂铁浓度的关系,线性拟合得到二者二级反应速率常数约为1.77×1010mol-1·dm3·s-1,接近扩散速率。溶液中有二茂铁存在时,400 nm的动力学曲线同样需要三指数函数拟合;随着二茂铁浓度增加,32M1NN衰减更快,25 μs成分的拟合指前因子更小。当溶液中二茂铁浓度为0.17 mmol/L时,32M1NN衰减时间常数仅为230 ns,而25 μs成分指前因子只有无二茂铁存在时的一半。这表明三重态猝灭剂二茂铁的存在使该物种的产率降低,说明32M1NN为该物种的前驱体。

图3 32M1NN在乙腈(Ar气氛)中600 nm处衰减速率常数与二茂铁浓度关系
2.2 瞬态物种指认
根据瞬态吸收光谱、二茂铁猝灭实验,以及邻硝基苯基类物质关于激发态分子内氢转移的研究报道[9-12],25 μs物种可能为32M1NN发生分子内氢转移后生成的顺式硝酸亚基中间体(以Z-aci-nitro表示,下同,结构见图4)。本文在TD-B3LYP/6-311++G(d,p)计算水平下得到Z-aci-nitro和分子内氢转移后双自由基的垂直激发能和振子强度,如表1所示。尽管Z-aci-nitro和双自由基结构在400 nm附近都有垂直跃迁,但前者的振子强度比后者大得多,比如Z-aci-nitro在392.2 nm振子强度为0.1161,而双自由基在402.1 nm振子强度只有0.0351。如果瞬态吸收实验中的25 μs物种是双自由基,则其衰减后应能看到400 nm附近光谱强度的增加,然而事实并非如此。25 μs物种不指认为双自由基的另一个理由是,其衰减时间常数并不受溶剂极性的影响。由此推测反应机理可能为:32M1NN的甲基氢原子转移到硝基生成双自由基中间体,接着发生较快的系间窜越过程生成Z-aci-nitro物种;Z-aci-nitro中间体不稳定,寿命约为25 μs,能异构化形成更稳定的E-aci-nitro异构体或发生其他反应而衰减。图4为用CASSCF/CASPT2方法和DFT方法计算的各个反应中间体的能量(图中用相对基态2M1NN的能量表示)。按照推测的反应机理,计算的反应中间体能量逐渐降低,计算的能量和反应历程吻合。
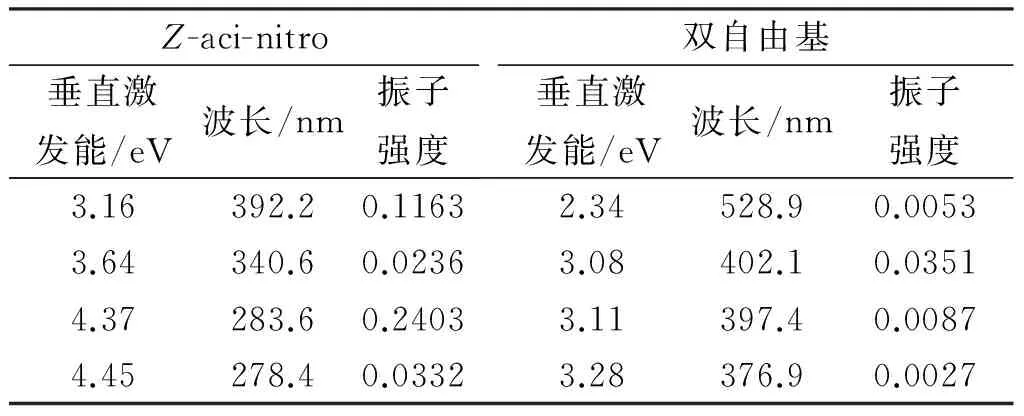
表1 在TD-B3LYP/6-311++G(d,p)计算水平下Z-aci-nitro和双自由基的垂直跃迁能与振子强度

a: CASSCF/CASPT2(10,10)/6-31G*,b:B3LYP/6-311++G(d,p),单位kcal/mol图4 计算的反应中间体能量示意图
2.3 反应机理
32M1NN的衰减时间常数约为800 ns,推测其分子内氢转移反应的速率常数约为1.0×106s-1,比邻硝基甲苯三重态分子内氢转移反应慢了三个数量级[12]。邻硝基甲苯T1态为3(n,π*),氧原子的一个n轨道电子跃迁到了π*轨道,此时氧原子的n轨道缺一个电子,氢原子能直接转移到氧上形成共价键,因此反应速度极快,可达109s-1数量级。而对于(π,π*)构型的32M1NN的氢转移反应,其机理可能是由于通过氢键发生的电子转移耦合质子转移[18-19];或是通过能级稍高的3(n,π*)三重态发生一步氢转移。前者反应速率可达107~109s-1,显然与本文的实验结果不符。CASSCF(10,10)/6-31G*计算结果显示,2M1NN的T2态能量比T1高26.5 kcal/mol,因此室温下通过T2发生一步氢转移也不是有效途径。
为了进一步研究32M1NN分子内氢转移机理,本文在CASSCF(10,10)/6-31G*水平下计算得到了T1一步氢转移反应的过渡态结构,如图5所示。硝基和萘环的二面角为18.20°,转移的氢原子距氧原子和碳原子分别为0.1200 nm和0.1276 nm,氢原子在碳、氧之间伸缩振动的虚频为4705 cm-1。该结构用CASPT2方法校正能量后得到相对于32M1NN的能量为9.44 kcal/mol。计算结果与32M1NN的衰减时间常数符合。
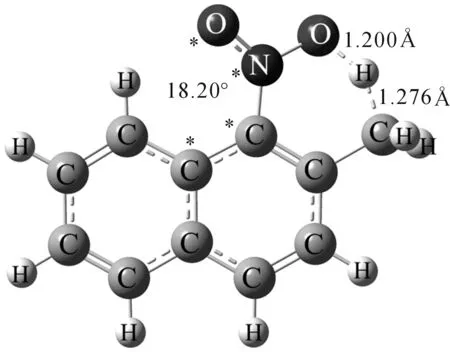
图5 在CASSCF(10,10)/6-31G*水平下算得分子内氢转移过渡态结构
三重态NPAHs也能发生分子间的氢转移反应,比如三重态硝基芘能夺取苯酚类物质羟基上的氢原子,并且利用瞬态吸收光谱可以观察到氢转移后生成的自由基[18-19]。但是32M1NN在甲醇和异丙醇溶剂中未观察到明显的分子间的氢转移反应,这表明分子内的氢转移速率较快,也证明32M1NN与溶剂分子难以形成有效的氢键通道,所以未能看到三重态2M1NN和醇类发生夺氢反应而生成的自由基。

a.2-甲基-1-硝基萘;b.1-硝基萘图6 基态的2M1NN和1NN紫外可见吸收光谱
在CASSCF(10,10)/6-31G*计算水平下优化得到2M1NN的基态结构,硝基与萘环二面角为68.02°,N=O键长为0.1206 nm,C-N键长为0.1458 nm。图6为基态的2M1NN和1NN紫外可见吸收光谱。TD-PBE0/6-311++G(d,p)计算水平下得到2M1NN在353.27 nm处有一振子强度为0.0424的吸收,与实验值较吻合。从实验与计算结果中看到,2M1NN的第一吸收带为S0→S1(π,π*)的跃迁吸收,摩尔吸光系数比1NN小四倍左右。这说明邻位甲基的引入使硝基与萘环的扭转角增加,破坏了硝基-萘环的(电子共轭体系,(π,π*)吸收带强度减弱。硝基与萘环较大的二面角使分子内氢转移成为可能。同时这种结构也利于产生芳氧自由基。
3 结 论
乙腈溶剂中激光光解2M1NN的瞬态吸收实验,直接观测到了衰减时间常数为25 μs、最大吸收峰在400 nm的瞬态物种。与量子化学计算预测的甲基氢分子内转移至硝基,形成的顺式硝酸亚基中间体吸收位置一致。二茂铁猝灭实验证实分子内氢转移发生于32M1NN。先生成双自由基中间体,再系间窜越生成Z-aci-nitro,Z-aci-nitro寿命约为25 μs。
[1] GIUSSANI A. Toward the understanding of the photophysics and photochemistry of 1-Nitronaphthalene under solar radiation: the first theoretical evidence of a photodegradation intramolecular rearrangement mechanism involving the triplet states[J]. Journal of Chemical Theory and Computation,2014,10(9):3987-3995.
[2] HEALY R M, CHEN Y, KOURTCHEV I, et al. Rapid formation of secondary organic aerosol from the photolysis of 1-Nitronaphthalene: role of Naphthoxy radical self-reaction[J]. Environmental Science &Technology,2012,46(21):11813-11820.
[3] ALBINET A, LEOZ-GARZIANDIA E, BUDZINSKI H, et al. Polycyclic aromatic hydrocarbons (PAHs), nitrated PAHs and oxygenated PAHs in ambient air of the Marseilles area (South of France): concentrations and sources[J]. Science of the Total Environment,2007,384(1):280-292.
[4] JARIYASOPIT N, MCINTOSH M, ZIMMERMANN K, et al. Novel nitro-PAH formation from heterogeneous reactions of PAHs with NO2, NO3/N2O5, and OH radicals: prediction, laboratory studies, and mutagenicity[J]. Environmental Science &Technology,2013,48(1):412-419.
[5] VOGT R A, CRESPO-HERNNDEZ C E. Conformational control in the population of the triplet state and photoreactivity of Nitronaphthalene derivatives[J]. The Journal of Physical Chemistry A,2013,117(51):14100-14108.
[6] LARSEN M A B, THΦGERSEN J, STEPHANSEN A B, et al. Transient IR spectroscopic observation of singlet and triplet states of 2-Nitrofluorene: revisiting the photophysics of Nitroaromatics[J]. The Journal of Physical Chemistry A,2016,120(1):28-35.
[7] HURLEY R, TESTA A C. Photochemical n→π* excitation of Nitrobenzene[J]. Journal of the American Chemical Society,1966,88(19):4330-4332.
[8] VOGT R A, REICHARDT C, CRESPO-HERNNDEZ C E. Excited-state dynamics in nitro-naphthalene derivatives: intersystem crossing to the triplet manifold in hundreds of femtoseconds[J]. The Journal of Physical Chemistry A,2013,117(30):6580-6588.
[9] SCHMIERER T, LAIMGRUBER S, HAISER K, et al. Femtosecond spectroscopy on the photochemistry of ortho-Nitrotoluene[J]. Physical Chemistry Chemical Physics,2010,12(48):15653-15664.
[10] SCHMIERER T, SCHREIER W J, KOLLER F O, et al. Impact of vibrational excitation on the kinetics of a nascent ketene[J]. Physical Chemistry Chemical Physics,2009,11(48):11596-11607.
[11] IL’ICHEV Y V, SCHWÖRER M A, WIRZ J. Photochemical reaction mechanisms of 2-Nitrobenzyl compounds: methyl ethers and caged ATP[J]. Journal of the American Chemical Society,2004,126(14):4581-4595.
[13] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09, Revision D.01[CP]. Gaussian, Inc.,Wallingford CT,2013.
[14] WERNER H J, KNOWLES P J, KNIZIA G, et al. Molpro, Version 2012.1, a package ofabinitioprograms[CP].
[15] GOHDO M, TAKAMASU T, WAKASA M. Photochemical primary process of photo-Fries rearrangement reaction of 1-Naphthyl acetate as studied by MFE probe[J]. Physical Chemistry Chemical Physics,2011,13(2):755-761.
[16] GRITSAN N P, TSENTALOVICH Y P, YURKOVSKAYA A V, et al. Laser flash photolysis and CIDNP studies of 1-Naphthyl acetate photo-fries rearrangement[J]. The Journal of Physical Chemistry,1996,100(11):4448-4458.
[17] REICHARDT C, VOGT R A, CRESPO-HERNNDEZ C E. On the origin of ultrafast nonradiative transitions in nitro-polycyclic aromatic hydrocarbons: Excited-state dynamics in 1-Nitronaphthalene[J]. The Journal of Chemical Physics,2009,131(22):224518.
[18] ARCE R, PINO E F, VALLE C, et al. Photophysics and photochemistry of 1-Nitropyrene[J]. The Journal of Physical Chemistry A,2008,112(41):10294-10304.
[19] ARCE R, PINO E F, VALLE C, et al. A comparative photophysical and photochemical study of Nitropyrene isomers occurring in the environment[J]. The Journal of Physical Chemistry A,2011,115(2):152-160.
(责任编辑: 唐志荣)
Investigation on Nanosecond Transient Absorption Spectroscopyand Theoretical Calculation of Excited-state IntramolecularHydrogen Transfer of 2-Methyl-1-Nitronaphthalene
GUAN Huaiyu, XUE Jiadan
(School of Sciences, Zhejiang Sci-Tech University, Hangzhou 310018, China)
Excited-state intramolecular hydrogen transfer of 2-Methyl-1-Nitronaphthalene (2M1NN) in organic solvent was investigated by nanosecond transient absorption spectroscopy, Density Functional Theory (DFT) and complete a ctive space self-consistent field (CASSCF). Transient absorption spectra and kinetic constants of 2M1NN were obtained, and 3 decay time constants were gained at 400 nm, in which the 25 μs component was assigned to the intermediate ofZ-aci-nitro isomer after intramolecular hydrogen transfer. The second-order quenching rate constant of 2M1NN triplet by ferrocene is 1.77×1010mol-1·dm3·s-1.The results of theoretical calculations and transient absorption experiments predicted the possible hydrogen transfer mechanism, and indicated intramolecular hydrogen transfer takes place at the lowest triplet state of 2M1NN, forming biradical species, then undergoing intersystem corssing.
nitro-polycyclic aromatic hydrocarbons; triplet state; nanosecond transient absorption; CASSCF; excited-state intramolecular hydrogen transfer
10.3969/j.issn.1673-3851.2017.09.022
2017-01-07 网络出版日期: 2017-03-28
浙江理工大学科研启动基金项目(1206841-Y);浙江省自然科学基金项目(LY16B030008);国家自然科学基金项目(21202032,21473163)
关怀昱(1991-),男,湖南长沙人,硕士研究生,主要从事光化学反应动力学方面的研究。
薛佳丹,E-mail:jenniexue@126.com
O644.12
A
1673- 3851 (2017) 05- 0742- 05
