复方中药国际化开发实例分析*
2017-08-31孙鹤郭治昕李凌艳章顺楠何毅马晓慧王根辈王平杨柳
孙鹤,郭治昕,李凌艳,章顺楠,何毅,马晓慧,王根辈,王平,杨柳
复方中药国际化开发实例分析*
孙鹤**,郭治昕,李凌艳,章顺楠,何毅,马晓慧,王根辈,王平,杨柳
(天士力控股集团/创新中药关键技术国家重点实验室天津300410)
复方中药作为植物药向美国食品药品监督管理局(U.S.Food and Drug Administration,FDA)申报新药是一项开拓工程、系统工程和创新工程,复方丹参滴丸是中国中药产品进行国际化开发的标杆及典范,是中国自主研发的第一个完成FDA国际多中心Ⅲ期临床试验的创新药,是全球第一个顺利完成FDA国际多中心Ⅲ期临床试验的复方中药产品。二十年磨一剑的复方丹参滴丸国际化历程,跌宕起伏,探索了一条中国中药产品进行国际化开发的道路,从中积累了大量宝贵的中医药国际化经验。
天士力在复方丹参滴丸FDA申报过程中,在符合FDA法规的同时也充分考虑到了复方中药的特点,明确了产品适应症,采用国际金标准科学、并创新的制定了临床试验方案,顺利完成了Ⅱ期探索性临床研究,成功获得了Ⅲ期临床试验SPA,并最终顺利完成了国际多中心Ⅲ期临床试验。申报过程中,天士力不断完善标准和路径,引入了多项创新,比如非线性多因素回归模型的应用、符合中药特点的临床试验设计、中药药物相互作用的鸡尾酒方法学研究、复方中药指纹图谱研究、复方中药质量一致性研究、生物效价研究以及数据统计方法的创新等。
复方丹参滴丸的国际化历程,是天士力与美国FDA共同创新现代中药标准、完善天然药物产业链GMP体系、突破技术瓶颈、创新法规路径和提升产品价值的持续创新和共同创造历史的过程,是中国国家力量的展现,是人类医药历史的进步。
复方中药复方丹参滴丸T89国际化FDA申报
复方丹参滴丸(Dantonic®,美国研发代号:T89)在美国FDA法规条件下的研发经历了将近10年的周期,其中引入了许多重大的科技创新,并获得一些关键问题的突破性进展,这是全球首个由中国自主研发的产品完成美国FDAⅢ期临床试验,也是首个获得美国FDAⅢ期临床试验阳性结果的复方中药,为一个新的治疗药物品种——复方植物药用于全球临床开拓了一条重要的、全新的道路[1,2]。
天士力集团根据产品特点,同时开展了复方丹参滴丸的FDA注册研发和丹参胶囊的欧盟注册研发工作,让现代中药能够以药品身份尽快进入欧美主流医药市场。多年来,复方丹参滴丸已在全球30个国家和地区以处方药、OTC药品、天然健康产品等多种身份销售(表1),这既是对国际市场的探索和推广,也为未来产品在欧美市场的营销储备了经验、影响与人力资源。
T89在美国FDA注册总体可分为三个阶段:①1996-2006年处在“不断探索、寻找路径”阶段,先后两次申请IND并获准;②2006-2010年是“构划路径、取得突破”阶段,启动并顺利完成Ⅱ期临床研究,在FDA申请并开展了了5个平行临床试验(表2);③2010年至今是“优化路径、创新标准”阶段,现已完成Ⅲ期临床研究。

表1 复方丹参滴丸海外注册和销售国家
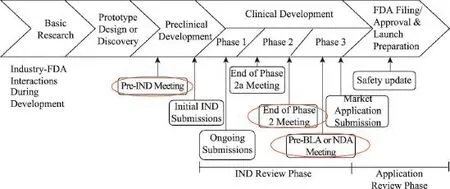
图1 新药研发各阶段FDA与申请者的沟通会议申请
1 T89临床研究实践
FDA与新药申请者正式的B类沟通会一般有三次:①临床申请递交前会议(Pre-IND会议);②Ⅱ期临床结题会议(EoP2会议);③新药申请生产递交前会议(Pre-NDA会议)。每次会议申请时间详见图1。
1.1 临床申请递交前(Pre-IND)会议[1]
Pre-IND会议主要讨论新药研发的全部流程,目的是促进申请者和新药审评部门的早期沟通,虽非强制步骤,但强烈建议进行。FDA鼓励申请者在新药开发过程的早期,在尚未积累足够临床申请数据前即同FDA进行沟通,以便申请者能在药学、临床、非临床开发过程中考虑FDA的专业建议,达成一定的共识。会议形式可以为电话会议或者面对面会议。
Pre-IND会议的流程是:①FDA会在接到申请者会议申请函的21天内确定会议日期;②申请者需要在开会的4周前递交更为详尽的会议资料;③申请者在开会前一周递交会议幻灯片;④会议资料发放至FDA多学科评审小组进行审评,之后FDA内部举行会议讨论,开会前FDA发出初步回复意见;⑤若申请者认为对初步回复意见不需要进行额外的讨论,申请者可取消会议,则初步回复意见为官方纪要;⑥若会议如期举行,申请者需要根据FDA的回复意见准备现场答辩资料并且在会议前一到两天将会议幻灯片交至FDA指定的项目经理(Consumer Safety Officer,CSO);⑦会议召开后30天内,FDA发出正式会议纪要作为官方纪要。Pre-IND会议资料包括首页函、申请表(FDA 1571表)、目录、所需咨询问题(分学科列举)、先前经验(包括其他国家上市及人用经验等)、药学资料、临床前研究资料、临床研究方案。一般情况下FDA有权不回答未列入会议资料中的问题。目前,中国药物研发申请FDA存在一项主要的问题,即企业开发新药时不草拟商品说明书,而是边做边写,建议与国内外专家和保险公司协商后草拟一份可能的商品说明书,相当于在研发前设计好蓝图,并在会议上就关键点征求FDA专业意见。
1.2 II期临床试验
复方中药可以部分参考FDA生物药的评价标准进行研究——均是一种混合物,均来自天然(培养液或提取液)[3,4]。Ⅱ期临床是探索性研究,FDA关注重点是产品的安全性。Ⅱ期临床试验方案的设计基于对法规、对产品的理解和信心以及大量基础研究和临床应用。复方中药Ⅱ期临床研究基于:①产品的理解及大量数据的积累,充分体现复方中药的特点和“起效缓慢、但作用持久”等现象;②国际药政法规的解读;③可被西方接受的适应症。
1.2.1 临床研究的关键:适应症的选择及有效性
心绞痛(Angina Pectoris)是由于心肌急剧的、暂时的供氧和需氧不平衡所引起的临床症状。其临床特征为阵发性前胸压榨样疼痛感觉,主要位于胸骨后部,可放射至心前区、左上肢、颈部、左肩部和后背部,常发生于劳累或情绪激动时,持续时间为数分钟,休息或用硝酸酯制剂后上述症状迅速消失。临床上将心绞痛分为稳定性心绞痛、不稳定性心绞痛和变异性心绞痛三种类型。其稳定性包含两方面的含义:其一是指病情稳定;其二是指冠状动脉粥样硬化斑块稳定,无溃疡破裂夹层及血栓形成等不稳定因素。稳定性心绞痛的病理基础是冠状动脉粥样硬化斑块所致的固定性狭窄。慢性稳定性心绞痛是指心绞痛发作的程度、频度、性质及诱发因素在数周内无显著变化。2012年,美国慢性心绞痛病人数约为820万,占该国人口总数的2.6%,且每年新增确诊病人数约56.5万。

表2 5个临床适应症与FDA探讨
治疗心绞痛通常分为三种方式:①扩张血管,扩张冠状动脉,增加心肌供血量,使用药物通常为硝酸酯类药物;②减少心肌耗氧量,常用药物为β受体阻断剂、钙通道阻滞剂等;③增加心肌能量代谢。但是,这三种治疗都存在或多或少的副作用或严重的临床缺陷,如硝酸酯类药物会随着时间的延长产生药物耐受性;β受体阻断剂会造成心率减慢,对于心率较低的病人不适用,而雷诺嗪有明显的对QT间隙延长和临床不适应(如药物相互作用)等局限使用,因此寻找更为安全有效的治疗药物是临床的迫切需求。
天士力提供了以下5个临床适应症与FDA探讨(表2),经反复商讨,FDA同意开展适应症1和2,结合产品特点和国内应用经验,企业最终选择适应症1即心绞痛开展海外研发临床研究。
1.2.2 Ⅱ期临床结题(EoP2)会议
Ⅱ期临床试验结束后,建议申请者与FDA开展一次EoP2会议,这为申请者和FDA审评部门提供了如下机会:①评价新药开发(包括Ⅱ期临床)的结果;②同申请者讨论后续研究的相关方案和计划;③识别安全性问题、科学问题和/或其他潜在问题,并且如有可能,尽可能在Ⅲ期临床启动前解决;④识别用于支持新药申请所需的其他信息;⑤可以是多学科的会议,或者单独召开CMC EoP2会议、临床EoP2会议等;⑥会议形式和会议流程同Pre-IND会议。
2010年7月22-23日,天士力同美国FDA召开了Ⅱ期结题会议,会议充分肯定了T89的Ⅱ期研究结果。T89美国Ⅱ期临床研究结果显示:①T89每日两次规则服用可以改善最大运动耐受时间(Total Exercise Duration,TED),该结果在临床、统计学上均具有显著性意义(P<0.05);②T89药效学有清晰的量-效关系;③其他反映复方中药特点的终点指标,如减少每周心绞痛发作频率、降低每周的硝酸酯类药物消耗,都一致具有临床及统计显著意义,并且遵循几乎相同的量-效规则;④每一类别的不良反应出现频率都非常低,类同安慰剂组。
T89同其他抗心绞痛药物在延长TED方面的比较:FDA批准的抗心绞痛药物均在超过安慰剂22-39 s之间,T89达到了42 s。FDA批准的其他药物还没有一个超过T89对TED延长的程度,T89具有针对多靶点的多种药理作用,可以作用于心脏、血管、血液和能量代谢,其中的有效成分可改善心肌能量供给,从而减少心肌的“饥饿感”,缓解疼痛;还可减轻血管内皮炎症、缩小血管中的斑块;在不改变心率的情况下降低血液粘稠度,增加血液流变动力学,起到活血化瘀的作用。
1.3 Ⅲ期临床研究
从药理学角度来看,T89将会在血液粘稠度改善、血液流变动力学提升、血管内皮斑块的减少,心肌能量代谢增加,冠状动脉流量的增长等多方面、多靶点同时起到重要的治疗效果,组方简单明快,君臣佐使的定位明确,比例合适,为取得重大的临床效果提供强有力的依据。
Ⅲ期临床是关键性研究,FDA关注点与Ⅱ期不同。Ⅲ期临床研究中,FDA一般要求进行两个相互验证的Ⅲ期临床研究;难度高,更侧重于设计的合理性、科学性和方案的系统性、逻辑性。复方中药Ⅲ期临床研究的关注点一般是:①安全性、有效性;②复方药物配伍合理性;③质量一致性;④临床价值③注:安全、有效、质量可控的药物不一定有临床价值,如使用困难、价格太高、顺应性差等等,都影响药物的临床价值。。

图2 T89 FDA-SPA会议
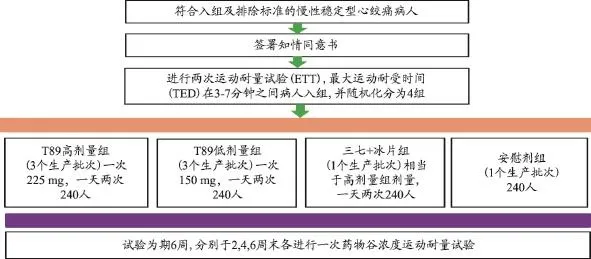
图3 T89Ⅲ期临床试验方案设计
1.3.1 同FDA就T89重大研究达成SPA共识
方案特殊评价(Special Protocol Assessment,SPA)是FDA与企业就重大或创新性研究设立的一个特别法规程序。FDA与企业达成SPA共识后,即成为双方共同遵守的约定,在新药评审(New Drug Application,NDA)时,将不再对达成共识的研究方案提出疑义。
鉴于T89Ⅱ期临床展示的优良临床疗效结果,预示着其在未来临床治疗上的重要价值。而且,这是首个复方处方中药在美国FDA法规下研发,组方和药理药效的复杂,很多问题必须及早确认。另外,根据天士力在该药物前期研究中所展现的严谨、细致的工作作风和企业信誉,在Ⅱ期临床结题会上双方达成了一致,FDA授予了T89的Ⅲ期临床SPA特许。
在SPA的原则下,我们根据不断深入研究所出现的问题、难度和复杂性,通过精心设计,严密组织,积极同FDA就Ⅲ期临床方案、药理致癌性研究和CMC研究启动了其他SPA程序的申请。通过广泛探讨,深入研究,最终同FDA达成多项具有里程碑式的重大共识,例如:①从原则上需要进行两个相互验证的Ⅲ期临床研究修订为只要一个Ⅲ期临床和两个在健康人群进行的小拆方研究;②从一般不要求多剂量多批次,到T89使用两个剂量及有差异的多批次用于Ⅲ期临床研究,通过临床研究疗效一致性证明质量一致性;也为CMC质量标准含量上下限设定提供了依据。
2012年4月26日,在美国FDA总部召开了SPA会议:将两个Ⅲ期临床研究,修订为一个Ⅲ期临床、两个在健康人群进行的小拆方研究(图2)。T89Ⅲ期临床试验主要终点是:采用国际通行的金标准及同安慰剂组比较,在第4周末药物谷浓度时最大运动耐受时间(TED)相对于基线变化(图3)。但同时天士力要求了增加第2和第6周的ETT观察,以考查TED随服药时间而改变的趋势。
值得提醒的是,两个关键性临床试验合并为一个关键性试验的意义:①患者数由2 000多人减少至960人,一方面说明T89Ⅱ期临床的结果使FDA树立了信心,另一方面说明FDA认可T89安全窗宽的事实;②直接节约大量研究成本;③降低了试验风险,减少了伦理审查委员会(Institutional Review Board,IRB)的审批难度;④避免了两个试验入组竞争;⑤缩短了研究周期。总而言之,临床设计是解决关键瓶颈的路径。
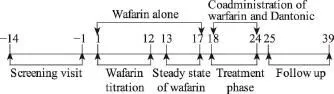
图4 T89与华法林药物相互作用临床研究

表3 服用T89前后INR均值

表4 T89对华法令药代动力学影响
1.3.2 临床方案设计的创新
在整个研发过程中,如何评价批次间一致性问题对临床结果的影响,如何能够更好的设计临床试验使得复方中药起效缓慢、作用持久的特点体现出来,如何能够真正使得临床研究的试验设计满足临床的治疗需求同时体现中医药的特色,如何解决复方中药组分配比的最佳比例,如何从中国人群使用剂量外推到海外不同种族人群的使用剂量,以及如何能够为了满足海外市场需求将剂量间隔适当延长提高顺应性等都给天士力的研发者们提出了一系列的挑战,而这些挑战的解决方案将会为中医药的国际化带来重要的指导作用。
综合运用了大量现代临床医学知识、转化医学方法、计量临床药理学技术、非线性、混合多因素模型量法等交叉学科的研究技术,模拟、分析、考察剂量、剂量间隔、给药时间、峰谷效应和病人群属分布参数的影响。通过优良的临床研究设计,体现了中药的特点,证实了中药治疗的高度:①中药能够挑战心血管临床试验的“金标准”(运动平板试验);②严格的入组/排除标准——背景用药:允许一种β受体阻滞剂或者钙离子通道抑制剂,不能服用长效硝酸甘油和/或雷诺嗪,短效硝酸甘油允许临时用于缓解症状。试验开始前7天和第0天两次基线运动耐量试验(Exercise Tolerance Test,ETT),两次ETT在3-7 min内并且差异小于15%。除两次基线ETT,Ⅱ期临床分别在第4周末和8周末各进行了两次分别在峰、谷浓度下的ETT。Ⅲ期临床在2、4、6周末的谷浓度时间各进行一次ETT。
1.4 中药的药物相互作用研究
在T89与华法林药物相互作用临床研究中,试验设计了一个开放的单次序交叉、健康受试者试验来评估T89对稳态华法林的药效学相互作用(图4)[5]。试验的首要终点为国际标准化比值(International Normalized Ratio,INR)在服用及没有服用T89情况下的变化,次要终点为安全性指标及华法林的药代动力学(Pharmacokinetics,PK)的变化。通过分析变量及计算可信区间(90%)以估计相互作用的程度。
试验结果显示,T89对达稳态华法林的PK/PD没有任何影响。PD统计分析结果表明,T89对已达到稳态华法林的PD也没有任何影响。安全性试验中没有出现任何SAE,所有AEs都很轻微,且与T89不相关;24个受试者中只有一例由于自身原因退出试验(表3、表4、图5)。
在过去的10年里,天士力集团秉承“勇于创新,勇于突破”的精神,在临床前试验、制剂开发与改进、临床试验设计与完成,以及数据统计管理等多个方面取得了一系列的突破性进展,这些工作具有里程碑式的重要意义。
T89的临床Ⅲ期试验表明:T89在主要临床终点上具有显著的量效关系、增加TED的作用明显优于安慰剂对照组和三七冰片拆方组。排除由于不可抗力(如国家战争、中心倒闭)导致研究数据不可采用的个别病例,对多种类型心电图异常的较严重稳定性心绞痛病人,在临床研究的前4周治疗期间,T89高、低剂量增加TED的作用类似;随后至第6周,高剂量组随服药时间而继续增加TED的速率保持不变,低剂量组的增速相对有所放缓,但均具统计学显著意义(P<0.001)且显著优于安慰剂对照组及三七冰片拆方组(P<0.05);第6周试验治疗结束时的点对点比较显示,高剂量组的作用高于低剂量组,且两组均显著优于安慰剂对照组和三七冰片拆方组(P<0.05)。
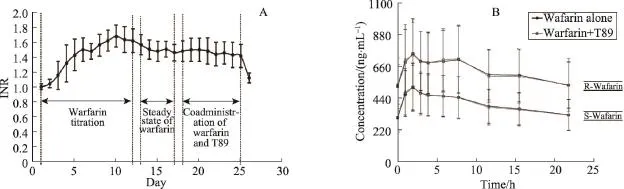
图5 INR变化曲线及T89对华法令PK影响曲线
次要疗效观察终点指标佐证主要临床终点指标,疗效证据成链。在4周治疗期后,T89高、低剂量组相对于安慰剂组可显著减少每双周硝酸甘油使用量,同比安慰剂组在此期间反而增加了硝酸甘油使用量。T89高、低剂量组相对于安慰剂组还可显著降低每双周心绞痛发作次数,同比安慰剂组几乎对心绞痛发作次数没有影响。
本试验首次采用大规模随机双盲国际多中心Ⅲ期临床试验的方法,通过T89高剂量组的疗效明显优于与其相当剂量的三七冰片拆方组的头对头比较,用临床研究解读了T89的组方基础,满足了对复方中药研发需进行拆方研究的药政管理要求。
鉴于试验用不同生产批次的T89在本试验的疗效上没有可见的差异,因此用于Ⅲ期临床的三个批次生产样品中的有效物质控制范围可作为上市产品质量标准依据。
本次试验再次证明了T89的临床安全性。整个试验期间没有发生任何与试验方案或T89相关的严重不良事件。所有其他一般不良事件均低频率、较轻微、可自愈,不同研究组之间的不良事件发生率没有区别。在已获得的临床前实验动物急毒、长期毒性实验等安全性证据和健康志愿者剂量爬坡临床安全药理证据之上,验证了T89在本试验使用的高、低剂量条件下临床使用安全。T89治疗慢性稳定性心绞痛的美国FDAⅡ期临床试验的安全性、有效性结果在Ⅲ期试验中得到了进一步验证。
2 T89药学④药学(Chemistry,Manufacturing and Controls,CMC)。研究实践
2.1 质量一致性问题
FDA在批准的植物处方新药绿茶提取物Veregen的植物学审评中指出“由于植物药允许以复杂的混合物形式存在,因此与非植物药相比,如何阐明质量的一致性是一个非常复杂的问题”;FDA和欧洲药品管理局(European Medicines Agency,EMA)均认可中药的活性物质为复杂的混合物,因此应采取多手段的质量控制策略来保证植物药的质量一致性[3,4,6,7]。
2.2 Ⅲ期临床样品要求问题
Ⅱ期临床样品需要符合研发需求,在符合GMP条件下生产;而Ⅲ期临床样品,对植物药来讲,FDA认为不能进行生物等效性评价,因此要求Ⅲ期临床样品的制备工艺应同拟上市产品一致,并且在GMP条件下生产。基于对Ⅱ期临床结果的信心,T89项目组在EoP2会议后全面实施了CMC工艺再提升、质量标准再完善,创新了许多新制备工艺、创造了新技术和新装备、优化了提取与制剂生产线、实施了厂房设备设施与工艺方法的全面再验证、提升了生产过程中的质量控制手段,完成了CMC的详细资料,递交FDA后,实施了Ⅲ期临床用药的生产。
2.2.1 CMC资料变更问题
除SPA会议等方式外,EoP2会议后,天士力还通过向FDA多次递交CMC变更资料,就T89项目开发进展中的众多关键问题进行了咨询,均取得FDA的书面回复。经多次沟通,在多项CMC关键问题上与FDA达成多项共识:①在提取物组方方面:在Ⅱ期临床研究的基础上,确定了以提取物作为活性整体,且作为产品处方原料,据此来描述处方设计、剂量设定等;并参照化学原料药(方式设定提取物质量标准,继续通过实施提取物批次搭配(混合)投料提高指标成分的控制精度,提高产品批间质量一致性。这些也是按照我国《中药注册管理补充规定》中允许提取物作为处方配伍和天士力在中国国内生产复方丹参的一贯方法和标准而进行的④。②CMC开发永远是一个逐步完善的过程,可以根据设备更新、研究进展、工业4.0概念的实施,逐步递交变更资料,建议在Ⅲ期临床开始前,申请者提交一次详细的CMC变更资料并获得FDA认可。在多轮沟通中,FDA逐渐接受了我们提出和实施的植物药质量控制标准参照中国中药研究相关技术要求、目前的技术路径及欧洲草药管理的相关要求来评价与实施。
2.2.2 化学成分差异问题
(1)美国药典USP<905>中的含量均匀度
FDA要求“重量差异应符合美国药典USP<905>要求”[3,4],如何实施这一要求,我们同FDA进行了如下讨论:USP<905>规定适用于化学药,以成分含量数据进行评价,对于T89来说,药用物质为丹参三七提取物的活性整体物质,提取物为复杂的混合物而非单独的化合物,采用提取物中某一种或某几种指标成分含量数据进行评价并不能代表T89提取物整体状态。因此,我们建议参照欧洲药典中胶囊剂项下规定进行检测(与《中国药典》要求一致)[6]。最后,以上这些讨论观点获得了FDA的认可。
(2)稳定性指标和显著性差异问题
ICH将药品含量稳定性的显著性差异定义为比初始值超出±5%。如果有显著性差异的,应进行趋势评价。由于植物药成分复杂,天士力参照欧盟指南(欧盟草药/传统药质量指南(EMA/CPMP/QWP/2819/ 00Rev.2)[6],对于治疗活性成分未知的植物药,经申请者评估,货架期成分含量变化显著性差异进行了一定自主标准的设定。FDA认可我们按欧盟稳定性评价要求实施,FDA同时建议我们继续积累稳定性数据,供递交新药申请时评价。在交流及沟通中发现,在植物药质量标准建立方面,全世界都处于一个探索及研究阶段,总体原则与中国食品药品监督管理总局相关要求一致;即强调安全性评价、质量一致性评价,并结合药理药效、临床样品设计及临床研究结果,确定标准的合理性。这给中国制药企业领导世界植物药研发提供了一个极好的发挥机会。
(3)安全性检测指标问题
药材标准要求与国内大致相同,除如性状、鉴别、含量测定等项目外,FDA强调重金属、黄曲霉素、农药残留、微生物等方面全面检验、检查。提取物标准也同样强调重金属、溶剂残留、农药残留、黄曲霉素、微生物等检查项目,并根据植物来源和药效作用,增加侧柏酮、硝酸盐等检测项目。
(4)质量一致性评价问题
药材基源需鉴定至种(及变种、亚种),强调产地、良好种植采收管理,药材处理、加工及储存要遵循GMP管理。根据工艺质量要求,强调批内均匀性、质量稳定性。对提取物及制剂,要严格执行美国FDA的cGMP要求,在厂房、设备设施、工艺质量设计及管理方面建立全面质量管理与风险评估。含量控制指标的建立与评价是逐步提升与优化的。T89在FDA法规下的海外研发实施的不同阶段,企业应保持与FDA的联系并共同探讨含量控制指标与评价的合理性;结合药理药效、临床样品设计及临床研究的结果,逐步提升与优化。
2.3 FDA一直关注并持续推动的问题
2.3.1 生物效价研究
与国内一致,对于药材、提取物、制剂的质量控制,FDA一直强调生物效价研究(Biological Assay)的重要性。2016年底,FDA最新颁布的植物药指南中再次推荐进行生物效价研究[5]。在与FDA沟通过程中,FDA在回复意见时期望天士力建立与药效相关的生物效价研究方法。为此,我们也针对T89尝试着建立了一些相应的生物效价研究方法。
T89研发团队结合T89的作用机理、药效特点探索开发了基于斑马鱼急性心肌缺血模型的T89生物效价方法。在斑马鱼急性心肌缺血模型的研究中,斑马鱼与人基因同源性达87%,试验周期1-3天,毫克级样品,多控板检测,费用为啮齿类1/10-1/100。
2.3.2 质量平衡问题
FDA建议提供化学标记物占提取物的重量百分比并对提取物中其他未明确的组分加以讨论。建议在上市前提取物和制剂的质量标准中,设定大类组分的标准用于阐明质量平衡。FDA一方面认可提取物活性整体的质量控制复杂,一方面又希望通过化学组分质量平衡、生物效价等研究来寻找到提取物活性整体的生物等效性评价方法,以方便产品生产过程质量控制评价以及上市后的变更评价。
2.4 小结
复方中药在FDA法规下的研发实践是一个不断深入、不断完善的过程,是与FDA不断沟通、不断理解的过程,更是一个双方共同全面创新、保证产品质量的系统工程:对欧美注册法规的深入理解、对产品质量属性的深入理解、对产品技术特点的深入理解、全面贯彻实施cGMP的质量管理,是不论产品国内上市还是国外上市的共同要求,也是天士力一直坚持的理念和做法。本文认为任何国内医药企业在准备进军海外前,一定要练好内功,提升标准,只有这样,才能对产品充满信心,才能把自己的实践与FDA分享并得到认可。现总结天士力的实践经验如下:
2.4.1 第一阶段工艺设计
Ⅱ期临床试验后,从工程项目交接、厂房设备验证、工艺技术转移与确认、到临床样品生产,全面实施了符合FDA标准的cGMP的质量管理。质量风险管理以“质量源于设计(QbD)”的理念,按照预定用途,控制关键控制点来开展的,以确保新工厂高效的运行;并保证有效实施工艺控制。该阶段主要的策略、方法包括:①物料质量控制策略:T89项目涉及的药材、辅料、内外包装材料以及工艺过程中所用溶剂和物料,基于风险评估基础上采用全面质量控制策略和专项工作开展,保障满足FDA法规要求;②设备选型及创新:为T89项目,我们共制定了用户需求文件(User Requirement Specifications,URS)一百多份,提取系统用户需求共计近一百项技术要点,浓缩系统用户需求共计50多项技术要点,T89项目多项技术与装备的国际化创新成果,经放大转移,已同时成功应用于了国内产品的提取、滴制等生产过程。
T89项目采用了高速滴制系统代替了传统的滴制方式,此外还采用了高速滴制系统深冷技术、超高速磁悬浮技术、高粘度液体无波动连续输送技术、高精度薄壁制管技术、超长行程在线清洗(Clean In Place,CIP)软管输送技术、高粘度丸剂快速干燥技术、快速包衣技术等等。从2010年,我们就开始对滴制工艺及设备进行了技术攻关,2012年2月全部验证工作完成。
2.4.2 第二阶段工艺验证实施
首先,按照产品生命周期设计进行验证,从“纸工厂”实现新车间建设运行,保证完成技术转移及工艺验证的顺利实施。T89生产车间建造按照“良好设备管理规范”(Good Equipment Practice,GEP)进行了全面质量管理。T89系统验证实施工作完成了近500个专项工作的集合,且项目要求工作标准和逻辑性进行严格控制。工艺验证遵循“质量源于设计”的理念,采用产品生命周期验证方法,关注工艺的合理性、可控性和稳定性。为确保Ⅲ期临床样品生产前工艺稳定,实施工艺设计和工艺确认结合的方式,通过多批试验和验证批次积累经验,找到关键质量属性(Critical Quality Attributes,CQA),确认可达到关键工艺参数(Critical Process Parameter,CPP)控制范围,达到验证要求。
以提取工艺验证研究为例,首先进行了工艺过程风险分析,确定关键质量属性(CQA)和关键工艺参数(CPP);其次确认关键工艺步骤;接着研究关键工艺参数范围(设计空间DOE),通过浓缩液密度、冷藏温度、等等因素影响研究,确认关键工艺控制参数;最后进行预验证与验证:通过验证取样、检测,确认关键工艺控制参数可控,符合T89提取物质量标准多项指标的限度要求。制剂工艺控制使得含量范围更加稳定。
2.4.3 第三阶段持续工艺确证
过程分析技术(Process Analytical Technology,PAT)的应用是总体质量控制的一部分,目标是对关键工艺步骤进行控制,将波动大的输入控制成稳定的输出,将产品的质量控制向上游转移,通过过程预防和持续性改善,保证最终产品的质量更均一、稳定。因此,CMC研究是基于质量风险管理,提倡质量源于设计、依靠技术创新、强化过程控制、实施全面验证、持续完善标准的系统工程。
3 T89在FDA法规下的海外研发体会与思考
T89海外研发项目存在10个方面的关键瓶颈,在研发初期,临床研究、CMC研究、保持与FDA的技术沟通尤为重要。在新药研发方面,FDA的要求主要体现在4个方面(这与国内一致):①研究科学化,强调真实性、可追溯性;②标准透明化,一方面遵循ICH、FDA、EMA、ISPE等完备的法规技术指南,另一方面强调根据研究项目具体情况合理评价;③强调安全性;④强调质量一致性。
复方中药新药申请对FDA是新的挑战,美国FDA对中药新药申请保持积极态度,既坚持原则又变通策略。需要申报方的勇于创新,同FDA经常互动,最终达成共识;复方中药的海外FDA法规下的临床研究、CMC研究、法规路径研究均是崭新的领域,是多学科的交叉体系,其过程创建了一种新模式,探索了一条新路径;研究过程是技术创新和标准升级的过程,是将从资源输出转变为技术价值输出的过程;中药新药研发应以满足临床需求为目标,临床效果为根本,用药安全为基础,以持续生产质量均一稳定的产品为要求,建立起研发新模式;从新药申请到文化认同是个漫长的过程、艰巨的工程,需要同仁们的共同努力,持续创新。
最后,关于中药研究与发展的三点思考和建议:首先,国内研究与注册审评应分类进行,促进中药的创新和国际化。譬如创新中药品种应从源头按国际化要求实施研究、评审,其它品种可按传统中药进行研究、评审;已上市品种可结合国家中药大品种二次开发专项研究来进行系统的提升;同时,对按国际化要求实施研究的品种应给与优质优价,增强企业持续发展的动力。其次,加快实现国际互认。对于已实施国际化研究的品种应建立回归国内的承接认可制度;实现中国中药研究与审评的国际认可,同时达到墙外开花香满园的效果。最后,国家通过中药品种的海外上市,展现国家科技力量,并逐步实现中药研发及审评技术标准的国际领导话语权。
致谢:感谢国家重大新药创制专项对本项目的支持!
1孙鹤,闫希军.现代中药国际化研究.北京:中国医药科技出版社, 2014.
2闫希军,孙鹤.现代中药国际化思考与实践.北京:中国医药科技出版社,2014.
3Food and Drug Administration.Guidance for Industry:Botanical drug products.Rockville,MD,U.S.A.,2004.
4Food and Drug Administration.Botanical Drug development:Guidance for Industry.Rockville,MD,U.S.A.,2015.
5Ping Wang,He Sun,Liu Yang,et al.Absence of an Effect of T89 on the Steady-State Pharmacokinetics and Pharmacodynamics of Warfarin in Healthy Volunteers.J Clin Pharmacol,2014,54(2):234-239.
6Guidance on Quality of Herbal Medicinal Products/Traditional Herbal Medicinal Products.European Medicines Agency,London,UK,2011.
7General guidelines for methodologies on research and evaluation of tra⁃ditional medicine.World Health Organization,Geneva,Switzerland, 2000.
Case Study of Compound Traditional Chinese Medicine Globalization
Sun He,Guo Zhixin,Li Lingyan,Zhang Shunnan,He Yi,Ma Xiaohui,Wang Genbei,Wang Ping,Yang Liu
(State Key Laboratory of Core Technology in Innovative Chinese Medicine Development, Tasly Group,Tianjin 300410,China)
Compound Traditional Chinese Medicine(CTCM)applying for FDA approval as botanical drug is a systemic and innovative work.Compound Danshen Dripping Pill(T89)is the first CTCM completed FDA global multi-center Phase III clinical trial,which has become a benchmark for TCM globalization.Extensive experiences have been accumulated through the 20 years’research and development.
Tasly fully considered the characteristics of CTCM while complying with FDA regulations during the entire application process.In which,product indication was clearly defined and agreed,clinical trial protocol was scientifically and innovatively designed with gold standard,phaseⅡstudy was well completed,SPA for phaseⅢstudy was obtained, and eventually the global multi-center phaseⅢstudy was successfully completed.In order to better support T89’s application,Tasly conducted series of additional studies and introduced many innovations,such as drug-drug interaction study for TCM,finger printing study for CTCM,quality consistency study,biological assay study and etc.
Globalization of T89 is not only a product creating history,but also a process for Tasly and the US FDA working together to continuously innovate and create history in innovating modern TCM standards,improving industrial chain GMP system for botanical drugs,making technical breakthroughs,exploring regulatory pathways and increasing product value.It represents the national power and historical progress in medicine.
Compound Traditional Chinese Medicine,Compound Danshen Dripping Pill,T89,globalization,FDA appli⁃cation
10.11842/wst.2017.06.006
R289
A
(责任编辑:马雅静,责任译审:王晶)
2017-03-20
修回日期:2017-06-20
*科学技术部国家“重大新药创制”科技重大专项(2008ZX09401-006):以现代中药为主,面向国际的创新药物研究与开发技术体系建设,负责人:闫希军。
**通讯作者:孙鹤,博士,天士力控股集团副总裁,创新中药关键技术国家重点实验主任,主要研究方向:创新药物研发。
⑤作者建议制定中国“提取物组方相关研究技术指南”。
