注射用亚叶酸钙与葡萄糖注射液、氯化钠注射液的配伍稳定性考察Δ
2017-08-22李婵艺白万军董占军河北省人民医院药学部石家庄05005广东药科大学中心实验室广州50000河北医科大学研究生学院石家庄05007
安 静,李婵艺,李 倩,白万军,董占军#(.河北省人民医院药学部,石家庄 05005;.广东药科大学中心实验室,广州 50000;.河北医科大学研究生学院,石家庄 05007)
·临床药学与研究·
注射用亚叶酸钙与葡萄糖注射液、氯化钠注射液的配伍稳定性考察Δ
安 静1*,李婵艺2,李 倩3,白万军1,董占军1#(1.河北省人民医院药学部,石家庄 050051;2.广东药科大学中心实验室,广州 510000;3.河北医科大学研究生学院,石家庄 050017)
目的:考察注射用亚叶酸钙与葡萄糖注射液、氯化钠注射液的配伍稳定性。方法:参照临床常用质量浓度,取注射用亚叶酸钙各3支(每支相当于亚叶酸钙100 mg),分别与葡萄糖注射液250 mL、氯化钠注射液250 mL配伍,在室温光照或避光条件下,分别于0、1、2、3、4、6、8、12、24、36、48 h考察各配伍液的外观,检测其pH值和不溶性微粒数,并采用高效液相色谱法测定各配伍液中亚叶酸钙的含量。结果:在上述条件下,各配伍液均未见颜色变化,也无气体、沉淀、浑浊等现象出现;pH值无明显变化(RSD<2%,n=11)。配制后0 h时,各配伍液中 10 μm的微粒数较多,但随着时间的延长,其数量有所下降,且48 h内各配伍液中 10 μm和25 μm的微粒数均符合2015年版《中国药典》的规定。在避光条件下,各配伍液中亚叶酸钙的相对百分含量变化不大(RSD<2%,n=11);而在光照条件下,各配伍液中亚叶酸钙的相对百分含量明显降低,分别降至94.5%(与葡萄糖注射液配伍)和88.4%(与氯化钠注射液配伍)。结论:注射用亚叶酸钙在葡萄糖注射液中更稳定,且光照条件可影响其配伍稳定性。注射用亚叶酸钙与葡萄糖注射液、氯化钠注射液配伍后,应避光保存,并尽快使用。
注射用亚叶酸钙;葡萄糖注射液;氯化钠注射液;配伍稳定性
《医疗机构药事管理暂行规定》[1]明确指出,医疗机构要根据临床需要逐步建立全肠外营养和肿瘤化疗药物等静脉药物调配中心(PIVAS),实行集中调配和供应。因此,越来越多的医院建立了PIVAS。通过实践表明,药物集中调配大大降低了药液被污染的几率,避免了开放式配制带来的微粒、热原等致害物的污染,从而避免了部分输液反应及不溶性微粒污染所致的潜在危害[2]。由于PIVAS的集中调配,配制后的成品输液由PIVAS配送至各病区需要一定的时间,且对于距离较远的科室,甚至可能需要数小时。在此过程中,由于药物分散至溶剂中,在溶剂、光照、温度等条件的影响下,药物形态、不溶性微粒等的变化情况均可影响输液的质量[3-10]。亚叶酸钙作为一种抗肿瘤辅助药物,主要用于大剂量甲氨蝶呤的解毒治疗,同时还可增强氟尿嘧啶的治疗效果。目前,关于亚叶酸钙与溶剂的配伍稳定性研究较少,且配伍后不溶性微粒数、pH值等的变化均未见相关报道[11]。因此,本研究在室温(25℃)光照或避光条件下,以配伍液外观、pH值、不溶性微粒数、含量变化等为指标,分别考察了注射用亚叶酸钙与葡萄糖注射液和氯化钠注射液配伍后48 h内的稳定性,以期为临床合理用药提供参考。
1 材料
1.1 仪器
2695型高效液相色谱(HPLC)仪、2487型紫外检测器(美国Waters公司);GWF-8JD型微粒分析仪(天津天河分析仪器有限公司);PHS-30型酸度计(上海雷磁仪器厂)。
1.2 药品与试剂
注射用亚叶酸钙(江苏恒瑞医药股份有限公司,批准文号:国药准字H32022391,批号:15121617,规格:按C20H23N7O7计算100 mg);葡萄糖注射液(华润双鹤药业股份有限公司,批准文号:国药准字H11020621,批号:H201511022,规格:250 mL∶12.5 g);氯化钠注射液(石家庄四药有限公司,批准文号:国药准字H13023201,批号:1510063702,规格:250 mL∶2.25 g)。
亚叶酸钙对照品(中国食品药品检定研究院,批号:100252-201204,纯度:99.9%);磷酸盐标准缓冲液(瑞士梅特勒托利多公司,pH为7.00);甲醇为色谱纯,水为纯净水,其余试剂均为分析纯。
2 方法与结果
2.1 色谱条件
参照2015年版《中国药典》(二部)[12],采用HPLC法测定配伍液中亚叶酸钙的含量。色谱柱:Hypersil ODS2(250 mm×4.6 mm,5 μm);流动相:含0.1%四丁基氢氧化铵的磷酸氢二钠缓冲液(取10%四丁基氢氧化铵水溶液8.0 mL和磷酸氢二钠2.2 g,加水溶解并稀释至780 mL,并用磷酸调节pH值至7.8)-甲醇(78∶22,V/V);流速:1.0 mL/min;检测波长:280 nm;进样量:20 μL。
2.2 溶液的配制
2.2.1 对照品贮备液 精密称取亚叶酸钙对照品适量,置于10 mL量瓶中,用水溶解并定容,得质量浓度约为1.0 mg/mL的对照品贮备液,备用。
2.2.2 供试品溶液 模拟临床常用质量浓度配制。精密称取注射用亚叶酸钙31.2 mg,置于25 mL量瓶中,用葡萄糖注射液溶解并定容,得质量浓度约为1.25 mg/mL的混合溶液。精密量取上述混合溶液1 mL,置于10 mL量瓶中,用水稀释至刻度,得亚叶酸钙质量浓度为125 μg/mL的供试品溶液(Ⅰ)。以氯化钠注射液为溶剂,同法配制得亚叶酸钙质量浓度为125 μg/mL的供试品溶液(Ⅱ)。
2.3 专属性考察
按“2.1”项下色谱条件,分别取水、葡萄糖注射液、氯化钠注射液、供试品溶液(Ⅰ)、供试品溶液(Ⅱ)各适量,进样分析,记录色谱图。结果表明,该方法专属性良好,注射液中的其他成分不干扰亚叶酸钙的测定,详见图1。

图1 高效液相色谱图Fig 1 HPLC chromatograms
2.4 标准曲线的绘制
精密量取对照品贮备液适量,用水依次稀释得质量浓度为15、30、60、120、160、200 μg/mL的对照品溶液,按“2.1”项下色谱条件进样分析,记录色谱图。以峰面积(A)为纵坐标、待测物质量浓度(c)为横坐标进行线性回归,得到回归方程为A=98 770.0c+505 461.8(r=0.999 3,n=5)。结果显示,亚叶酸钙质量浓度在15~200 μg/mL范围内线性关系良好。
2.5 精密度试验
分别配制低、中、高质量浓度(30、120、160 μg/mL)的亚叶酸钙对照品溶液,连续进样5次,记录峰面积,考察日内精密度;连续测定3 d,根据当日标准曲线计算其实测质量浓度,考察日间精密度。结果显示,日内RSD分别为0.6%、0.7%、1.0%(n=5),日间RSD分别为1.5%、0.2%、1.2%(n=3),表明精密度良好。
2.6 回收率试验
2.6.1 亚叶酸钙在葡萄糖注射液中的回收率 精密量取质量浓度分别为30、120、200 μg/mL的亚叶酸钙对照品溶液各2.5 mL,分别置于5 mL量瓶中,各加入供试品溶液(Ⅰ)2.5 mL,用水定容,摇匀,各质量浓度平行配制3份,进样测定,计算回收率。结果显示,各样品的平均回收率分别为101.6%、101.1%、104.0%,RSD分别为1.2%、1.4%、1.2%(n=3)。
2.6.2 亚叶酸钙在氯化钠注射液中的回收率 精密量取质量浓度分别为30、120、200 μg/mL的亚叶酸钙对照品溶液各2.5 mL,分别置于5 mL量瓶中,各加入供试品溶液(Ⅱ)2.5 mL,用水定容,摇匀,各质量浓度平行配制3份,进样测定,计算回收率。结果显示,各样品的平均回收率分别为99.7%、104.8%、97.6%,RSD分别为1.5%、1.7%、2.5%(n=3)。
2.7 稳定性试验
分别配制低、中、高质量浓度(30、120、160 μg/mL)的亚叶酸钙对照品溶液,置于进样器中(4℃),于48 h内每间隔2 h进样分析,记录峰面积。结果显示,亚叶酸钙峰面积的RSD分别为1.4%、0.3%、0.5%(n=25),表明其在48 h内稳定性良好。
2.8 配伍稳定性考察
按照临床常用质量浓度,在PIVAS配制。取注射用亚叶酸钙各3支,分别加入至葡萄糖注射液250 mL和氯化钠注射液250 mL中,得亚叶酸钙-葡萄糖配伍液和亚叶酸钙-氯化钠配伍液(亚叶酸钙质量浓度约为1.2 mg/mL)各2份,在室温光照或避光条件下,分别于配制后0、1、2、3、4、6、8、12、24、36、48 h考察各配伍液的外观、pH值、不溶性微粒数和亚叶酸钙的含量。
2.8.1 外观和pH值 分别于上述时间点在白色背景下观察各配伍液的外观,采用酸度计测定其pH值(玻璃电极为指示电极、饱和甘汞电极为参比电极。测定前,采用磷酸盐标准缓冲液校正仪器;测定时,应先用纯化水充分洗涤电极,然后将水吸尽后,再用供试品溶液充分洗涤,直至pH值的读数在1 min内变化不超过0.1为止)。结果显示,在室温光照或避光条件下,配制后48 h内各配伍液均未见颜色变化,也无气体、沉淀、浑浊等现象产生;pH值亦无明显变化,RSD<2%(n=11),详见表1。

表1 各配伍液的pH值Tab 1 pH value of mixtures
2.8.2 不溶性微粒 参照2015年版《中国药典》(四部)通则中的“不溶性微粒检查法”,采用光阻法于上述各时间点检测各配伍液中的不溶性微粒数[标示装量为100 mL以下的静脉用注射液、静脉注射用无菌粉末、注射用浓溶液及供注射用无菌原料药,除另有规定外,每个供试品容器(份)中含10 μm及10 μm以上( 10 μm)的微粒数不得超过6 000粒,含25 μm及25 μm以上(25 μm)的微粒数不得超过600粒][13]。用纯净水将供试品容器(250 mL)的外壁洗净,取配伍液各20 mL,采用微粒分析仪检测。各样品连续测定3次,弃去第一次测得的数据,取后续测定数据的平均值作为检测结果。结果显示,配制后0 h时,各配伍液中 10 μm的微粒数较多,但随着时间的延长,其数量有所下降;配制后48 h内,各配伍液中 10 μm和 25 μm的微粒数均符合药典规定,详见表2。
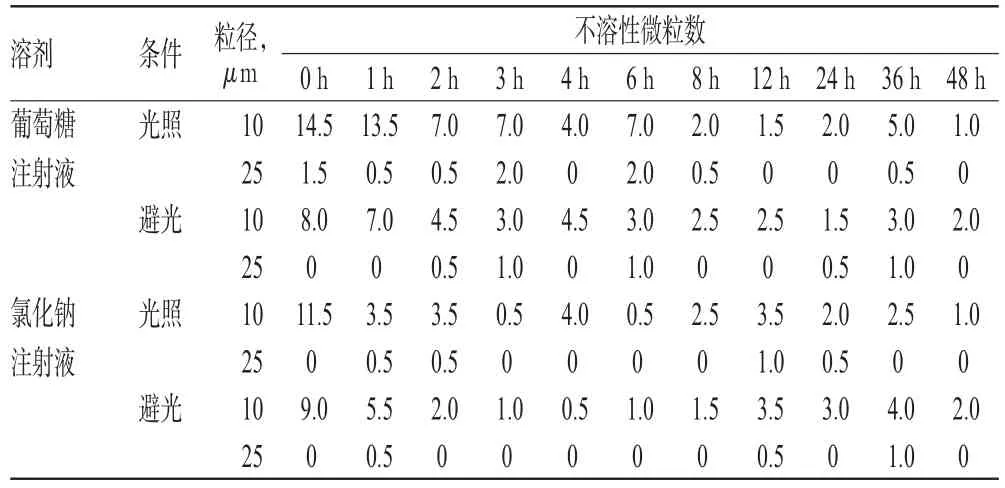
表2 各配伍液中的不溶性微粒数(粒/mL)Tab 2 The number of insoluble particles in mixtures(grain/mL)
2.8.3 相对百分含量 于上述各时间点取配伍液各适量,用纯净水稀释10倍,配制成质量浓度约为120 μg/mL的样品溶液,精密吸取20 μL,按“2.1”项下色谱条件进样分析,测定各配伍液中亚叶酸钙的质量浓度。以0 h时的质量浓度为100%,计算其他各个时间点的相对百分含量。结果显示,在室温避光条件下,48 h内各配伍液中亚叶酸钙的相对百分含量变化不大,RSD<2%(n=11);而在室温光照条件下,各配伍液中亚叶酸钙的相对百分含量明显降低,分别降至94.5%(与葡萄糖注射液配伍)和88.4%(与氯化钠注射液配伍),详见表3。
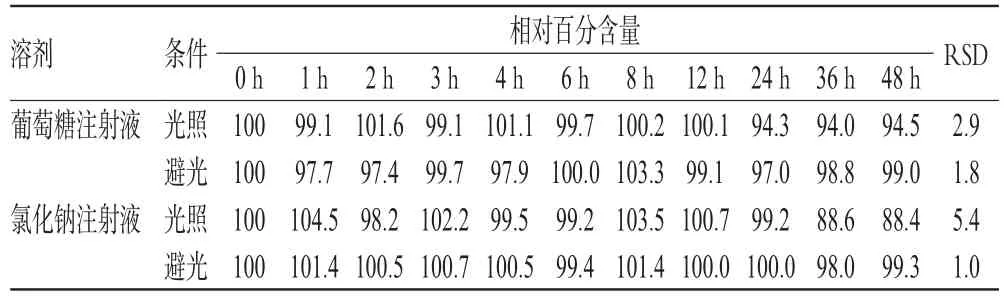
表3 各配伍液中亚叶酸钙的相对百分含量(%%)Tab 3 Relative contents of calcium folinate in mixtures(%%)
3 讨论
3.1 色谱条件的选择
亚叶酸钙的分子结构中含有氨基和羧基,为两性化合物,流动相的酸碱性对其峰形影响较大。前期研究中,本课题组参照文献[14]的方法,采用乙腈-10 mmol/L醋酸钠与三乙胺缓冲液的混合溶液(冰醋酸调pH值至5.5)(4∶96,V/V)为流动相,在286 nm检测波长下进行测定,但所得色谱峰峰形较差。最后,本课题组参照了2015年版《中国药典》(二部)的方法,在水相中加入了离子对试剂四丁基氢氧化铵,该离子对试剂可与亚叶酸钙形成离子对,大大改善了后者的峰形,且重现性良好。
3.2 不溶性微粒检测的注意事项
不溶性微粒的检测结果显示,在配伍液配制完成初期(配制后0 h),各配伍液中 10 μm的微粒数较多,但经长时间放置后其数量有所减少。可能是由于刚配制完成时,配伍液中存在大量的气泡,微粒分析仪在检测时会将气泡误读为微粒,使得微粒检测结果偏高。因此,在测定前应静置2 min消除气泡。
本研究在PIVAS洁净台上进行,尽可能减少了环境污染对检测结果的影响。若条件有限,无法到达洁净度要求,在试验过程中则应注意关闭空调、通风系统,避免人员随意进出,避免交谈;在将配伍液转移至供试品容器(测试杯)之前,还应用纯净水将容器外壁和注射器洗净,以免器具污染导致检测结果的不准确。除此之外,不适当的药物配伍、过多的药物联用、针头穿刺胶塞、安瓿切割等因素也会影响配伍液中的不溶性微粒数[15]。
3.3 光照对亚叶酸钙配伍稳定性的影响
相对百分含量测定结果显示,在避光条件下,注射用亚叶酸钙与葡萄糖注射液、氯化钠注射液配伍后,在48 h内保持稳定。而在光照条件下,注射用亚叶酸钙与葡萄糖注射液配伍后,在12 h内保持稳定,24 h时相对百分含量降至95%以下;注射用亚叶酸钙与氯化钠注射液配伍后,在24 h内保持稳定,36 h时相对百分含量降至90%以下,提示注射用亚叶酸钙在避光条件下较光照条件更为稳定。可能是因为亚叶酸钙在水溶液环境中,在多种离子的共同作用下,会发生缓慢的降解,而光照可加速其降解过程[16];此外,由于亚叶酸钙是非典型的两性化合物,阴离子结构中含有1个氨基和2个羧基,当其与溶剂配伍时,溶剂的酸碱性可影响亚叶酸钙的分子形态,弱酸性溶液有利于抑制亚叶酸钙分子的解离,提高其分子形式的比例[14],故与氯化钠注射液比较,亚叶酸钙在葡萄糖注射液中更为稳定。本研究结果表明,光照条件对注射用亚叶酸钙与2种溶剂的配伍稳定性有所影响。因此,在实际工作中,注射用亚叶酸钙与葡萄糖注射液、氯化钠注射液配伍后,应避光保存,并尽快使用。
本研究分别在室温光照或避光条件下,以外观、pH值、不溶性微粒、亚叶酸钙含量作为参考指标,对注射用亚叶酸钙与葡萄糖注射液、氯化钠注射液的配伍稳定性进行了考察,但温度、微生物、其他溶剂、不同输液袋材质等因素也可能影响其配伍稳定性,故有待于进一步的研究。
[1] 卫生部,国家中医药管理局,总后勤部卫生部.关于印发《医疗机构药事管理规定》的通知[S].2011-01-30.
[2] 杨晶晶,魏远明.静脉药物配置中心的管理与体会[J].海峡药学,2004,16(5):194-196.
[3] 黄丽君,刘炜,李力.托烷司琼注射液常见临床配伍稳定性考察[J].中国现代应用药学,2015,32(5):595-598.
[4] 王成湖,易林高.注射用奥扎格雷钠与含果糖注射液的配伍稳定性考察[J].中国药房,2016,27(2):191-192.
[5] 魏秀美.盐酸左氧氟沙星与卡络磺钠在0.9%氯化钠注射液中的稳定性考察[J].中国药房,2016,27(12):1671-1673.
[6] Mehta AM,van den Hoven JM,Rosing H,et al.Stability of oxaliplatin in chloride-containing carrier solutions used in hyperthermic intraperitoneal chemotherapy[J].Int J Pharm,2015,479(1):23-27.
[7] Trojniak MP,Mazzi U,Palozzo AC,et al.Stability of lyophilised oxaliplatin formulation in polyolefin infusion bags containing 5%dextrose injection[J].Eur J Hosp Pharm,2014,doi:10.1136/ejhpharm-2013-000323.
[8] 乔丽曼,黄晨,张慧,等.临床输注条件下核黄素磷酸钠的光稳定性研究[J].医药导报,2011,30(8):1101-1103.
[9] 张洁,赵金侠.氟脲苷在氯化钠和葡萄糖注射液中的稳定性考察[J].中国药师,2012,15(7):1002-1004.
[10] 李晓光,翟所迪,李珍,等.3种注射用盐酸表柔比星溶液的稳定性考察[J].中国药学杂志,2013,48(8):615-620.
[11] Karbownik A,Szałek E,Urjasz H,et al.Sability of calcium folinate(TEVA)in concentrate after re-use and in dilute infusions in 0.9%NaCl in polyethylene bags[J].Acta Pol Pharm,2013,70(2):301-307.
[12] 国家药典委员会.中华人民共和国药典:二部[S].2015年版.北京:中国医药科技出版社,2015:341.
[13] 国家药典委员会.中华人民共和国药典:四部[S].2015年版.北京:中国医药科技出版社,2015:114-115.
[14] 郑璐侠,段更利,高敏洁,等.反相高效液相色谱法测定亚叶酸钙制剂的含量及其稳定性考察[J].中国新药与临床杂志,2003,22(2):74-77.
[15] 石崇爱,陈凤莲,詹月敏,等.药物配置过程中不溶性微粒的来源分析及控制措施[J].海峡药学,2016,28(10):224-225.
[16] 湛常娟.注射用左亚叶酸钙制剂工艺及质量研究[D].合肥:安徽中医药大学,2013.
Study on Compatible Stability of Calcium Folinate for Injection Mixed with Glucose Injection and Sodium Chloride Injection
AN Jing1,LI Chanyi2,LI Qian3,BAI Wanjun1,DONG Zhanjun1(1.Dept.of Pharmacy,Hebei Provincial People’s Hospital,Shijiazhuang 050051,China;2.Central Laboratory,Guandong Pharmaceutical University,Guangzhou 510000,China;3.Graduate School,Hebei Medical University,Shijiazhuang 050017,China)
OBJECTIVE:To investigate the compatible stability of Calcium folinate for injection mixed with Glucose injection and Sodium chloride injection.METHODS:Referring to clinical common concentration,each 3 Calcium folinate for injection(each injection was equal to calcium folinate 100 mg)were respectively mixed with Glucose injection 250 mL or Sodium chloride injection 250 mL.At room temperature,under light or dark condition,the appearance of mixtures,pH value and the number of insoluble particles were investigated 0,1,2,3,4,6,8,12,24,36,48 h.The contents of calcium folinate in mixtures were determined by HPLC.RESULTS:Under above condition,the color of the mixtures had no change,and no gas,precipitation and turbidity was found;there was no evident change in pH values(RSD<2%,n=11).0 h after mixing,there was large number of particles 10 μm in mixtures,but the number of particle was decreased as time;within 48 h,the number of particles 10 μm and 25 μm in mixtures were all in line with the standard of Chinese Pharmacopeia(2015 edition).Under the protection from light condition,relative contents of calcium folinate in mixtures had no significant change(RSD<2%,n=11).Under light condition,relative contents of calcium folinate in mixtures decreased significantly,decreasing to 94.5%(mixed with Glucose injection)and 88.4%(mixed with Sodium chloride injection).CONCLUSIONS:Calcium folinate for injection is more stable in Glucose injection,and the stability of compatibility can be affected by light conditions.After mixed with Glucose injection and Sodium chloride injection,Calcium folinate for injection should be kept away from light and used as soon as possible
Calcium folinate for injection;Glucose injection;Sodium chloride injection;Compatible stability
R917;R969.3
A
1001-0408(2017)20-2764-04
2016-07-29
2017-05-17)
(编辑:张元媛)
河北省医学科学研究重点课题计划(No.20160062);河北省政府资助临床医学优秀人才培养项目(No.361003)
*主管药师。研究方向:临床药学。电话:0311-85988998。E-mail:anjingyaofen@163.com
#通信作者:主任药师,硕士生导师。研究方向:医院药学。电话:0311-85988604。E-mail:13313213656@126.com
DOI10.6039/j.issn.1001-0408.2017.20.08
