嵌段共聚物自组装中聚合诱导自组装方法的研究进展
2017-08-15孙汝柳
孙汝柳
(中国石化 北京化工研究院,北京 100013)
嵌段共聚物自组装中聚合诱导自组装方法的研究进展
孙汝柳
(中国石化 北京化工研究院,北京 100013)
综述了一种嵌段聚合物纳米微球的制备新方法——聚合诱导自组装(PISA)方法,该方法结合了活性聚合和自组装两个概念,可控制嵌段聚合物纳米微球的大小、形貌和表面性能,讨论了PISA方法的优势和缺陷,并展望了PISA方法的未来发展方向。
嵌段共聚物;自组装;活性聚合
嵌段共聚物的自组装行为是高分子领域的研究热点[1-2],通过嵌段高分子共聚物制备的组装体尺寸一般在10~1 000 nm,可极大地增加材料的物理、化学、生物等性能,在生物医药[3],催化[4]等领域应用价值巨大。通过自组装,嵌段共聚物可组装成球状、柱状、囊泡状等多种有序结构,且自组装结构可通过改变溶剂,相对分子质量等条件进行调节[5]。但嵌段共聚物自组装步骤通常比较繁琐,一般具有“聚合物合成-纯化分离-自组装-交联”四个步骤[6]。如何方便大规模地使用嵌段共聚物自组装制备纳米微球是工业上更为关心的研究课题。
为了克服嵌段高分子自组装步骤繁琐、无法大规模生产的缺点,Wang等[7-8]结合活性聚合和嵌段聚合物自组装的概念,一步法制备了具有核-壳结构的苯乙烯-丁二烯的纳米微球,这种方法被称为聚合诱导自组装(PISA)。使用PISA方法,可合成球形[9]、囊泡[10]、柱状[11]等各种拓扑结构的聚合物纳米材料[12-15]。随着活性自由基聚合的发展,PISA方法得到进一步发展,可使用各种官能性单体,得到各种功能化聚合物微球,制备聚合物纳米材料,可实现高固含量生产[14]。
本文介绍了采用结合活性聚合和自组装两个概念的PISA方法,控制嵌段聚合物纳米微球的大小、形貌和表面性能。PISA方法操作简单,适用于工业化生产,并展望了该方法未来的发展方向。
1 阴离子PISA方法
利用活性聚合方法,选择具有选择性的溶剂作为聚合溶剂制备聚合物纳米材料,以溶解性良好的第一链段为大分子引发剂,引发不溶的第二链段单体,制备AB嵌段聚合物,利用溶剂的选择性在聚合过程中诱导嵌段聚合物自组装。
Wang等[7]在正己烷中使用阴离子聚合聚苯乙烯(PS)-聚丁二烯嵌段聚合物,首次使用阴离子聚合方法实现了PISA。首先在正己烷中聚合丁二烯单体,得到聚丁二烯嵌段,正己烷为聚丁二烯的良溶剂,溶液体系这时表现为均相溶液。丁二烯聚合结束后,加入苯乙烯单体,得到两嵌段聚合物(PI-b-PS),由于正己烷是PS链段的不良溶剂,因此随着聚合的进行,嵌段聚合物开始自组装形成球状胶束。为了使得到的聚合物微球可进一步工业应用,又加入二乙烯基苯交联固定得到的聚合物微球。在机理上,Wang等[7]发现出现嵌段聚合物发生自组装之后,聚合速率出现明显的下降。
PISA方法结合了活性聚合和自组装两者的优点,通过改变嵌段聚合物的相对分子质量、两嵌段的比例、聚合物浓度、溶剂的选择性等调节所得聚合物微球的结构和性能。Wang等[7]发现在固定嵌段聚合物相对分子质量的情况下,改变嵌段聚合物的浓度可实现聚合物微球形貌发生从球状到椭球状再到柱状的转变。通过控制胶束的浓度,改变嵌段聚合物的相对分子质量,也可实现胶束形貌的改变。固定聚合物的浓度为15%(x),当嵌段聚合物的相对分子质量小于40 000时,聚合物以胶束为主。当嵌段聚合物的相对分子质量超过60 000时,聚合物胶束变为椭球形。进一步增加嵌段聚合物的相对分子质量到大于120 000时,聚合物转变为柱状。
Wang等[7]使用PISA方法制备的PI-b-PS聚合物微球,方法简单,可大规模应用,且可进一步改变微球的性质(如通过氧化水解改变聚合物微球的亲疏水性质,并用于负载无机纳米颗粒等)。此外该方法还可用于合成中空微球、两面微球、珍珠链状微球等结构。在聚合的过程中,加入PS均聚物,在嵌段聚合物发生自组装的情况下,PS均聚物被包裹在胶束内部。胶束交联固定之后,提取出PS均聚物,就可得到内部中空的微球。
2 活性自由基PISA方法
Wang等[7]使用阴离子聚合方法制备嵌段高分子组装体,并在橡胶轮胎、油墨等领域得到了应用。阴离子聚合方法对聚合条件要求苛刻,适用的单体种类和溶剂少,对水、醇、酸酐等活性基团敏感,这在一定程度上限制了PISA方法制备功能性材料。活性自由基方法克服了以上缺点,可扩大单体范围,更容易在聚合物上结合功能性单体,增加聚合物微球的功能和性质。
Ostu等[16]通过对二硫代氨基甲酸酯化合物调控自由基聚合的研究提出了“iniferter”的概念,这成为第一个真正意义上的活性自由基聚合。在这之后,活性/可控自由基聚合成为高分子化学研究中飞速发展的领域之一。至今,高分子化学家发现了多种活性自由基聚合方法,包括稳定自由基聚合(SFRP或NMP)[17-18],原子转移自由基聚合(ATRP)[19],可逆加成断裂链转移聚合(RAFT[20-21]或MADIX[22-24])等。
Ji等[25]使用RAFT方法研究了苯乙烯和马来酸酐在选择性溶剂中的共聚行为,并制备得到了具有核壳结构的聚合物纳米微球。通过RAFT方法PS和马来酸酐可一步得到嵌段聚合物P(S-alt-MAn)-b-PS,具有方法简单、容易操作的特点,且马来酸酐具有酸酐基团,易于修饰,可用于多种功能化。选择PS的不良溶剂乙腈作为聚合溶剂,苯乙烯和马来酸酐共聚速率远大于苯乙烯的均聚速率,因此聚合初期只能得到P(S-alt-MAn)共聚物,随着聚合的进行,得到了PS链段。由于PS链段在乙腈中的溶解性较差,因此在聚合过程中嵌段聚合物在乙腈中发生自组装行为,形成了P(S-alt-MAn)为壳,PS为核的纳米微球。通过进一步加入二乙烯基苯交联PS链段,还可得到稳定的纳米材料。
Zheng等[26]考察了溶剂聚合动力学的影响,并对聚合物的形貌进行了详细的表征。对比了在四氢呋喃(THF)和环己烷中PS大分子链转移剂引发4-乙烯基吡啶(4VP)聚合的聚合动力学行为,发现在良溶剂THF中,单体4VP转化率随着时间线性增加,而在聚4VP的不良溶剂环己烷中,单体4VP转化率则出现了一个转折点。聚合5 h后,聚合速率从0.164 mol/(L·h)突然降低到0.002 4 mol/(L·h),该转变表明在聚合体系中出现了聚合机理的转变。表征结果显示,这是由于出现了嵌段聚合物的自组装,形成了聚4VP核,导致单体聚合速率下降。
活性自由基聚合可用于PISA方法制备纳米微球,这样就增加了单体和聚合方法的选择范围。使用活性自由基聚合方法实现PISA,可使用ATRP[27],NMP[28],RAFT[29]等不同的受控游离基聚合(CRP)方法应用到更多的溶剂体系,如水、极性溶剂(醇)、非极性溶剂(烷烃)、离子液体[30]、超临界二氧化碳[31]等,可聚合功能性单体(如酸、醇、酰胺、盐等),可达到较高的固含量(w)(25%~50%)[32]。
嵌段共聚物自组装的形态是由体系自由能决定的,而体系自由能又由成核嵌段的伸展程度、壳嵌段的相互斥力和界面自由能决定。改变三种因素之间的平衡,就可达到调节聚集体形貌的目的,如调节嵌段共聚物的相对分子质量[33]、结构[34]、溶剂的组成和性质[35]、离子强度[36]及温度[37]等,就可得到球状、蠕虫状,棒状、囊泡、管状和复合胶束等丰富的形貌。
在PISA方法制备纳米微球的过程中,随着聚合时间的增加,嵌段聚合物B链段的聚合度不断增加,因此在聚合过程中得到的聚合物纳米微球会出现形貌的转变[38],这样就可在聚合过程中精确地控制微球结构。实际上从2009年以来,已经有几个研究组[39-41]证明了,在聚合过程中使用PISA方法,可得到不同形貌的聚集体。
Andreas等[42]使用含磷的单体甲基丙烯酸羟甲基磷酸二甲酯(PMP)为A链段,以丙烯酸苄酯为B链段,以甲醇为选择性溶剂,使用PISA方法,通过改变聚合物的组成,制备了不同形貌的纳米聚合物。图1为PMPx-PBzMAy在甲醇中的PISA的相图。由图1可知,在此体系中,AB两链段的长度都对最终聚合物的形貌有着重要的影响。当A链段较长时,A链段作为壳层,相互的排斥作用较大,因此限制了微球直接的相互融合。因而随着B链段聚合度的增加,聚合物自组装形貌并不出现变化,只是单纯的半径增加(16~76 nm)。而当降低A链段的聚合度时,聚合物微球会发生融合,导致球状形貌的不稳定,出现球-柱-囊泡的转变,如固定A的聚合度为32,随着聚合反应的进行,链段B的长度增加,形貌出现了球-球/柱混合物-柱-囊泡的变化。而进一步降低A链段的聚合度,则不会出现柱状结构。当亲溶剂链段A的长度较大时,由于链段之间的相互排斥作用较大,限制嵌段自组装体的融合,所以较难出现其他形貌聚集体。但是随着链段B长度的增加,聚合物微球的尺寸不断增加,相互作用增加,会降低壳层中A的链段密度,降低相互排斥,进而出现融合,这样就进一步增加了B链段的长度,仍然会出现球-线结构的转变。Zhang等[43]使用PISA研究了聚甲基丙烯酸N,N-二甲基氨基乙酯(PDMAEMA)和PS在甲醇中的聚合行为。选择了不同链长的PDMAEMAx(x = 22,36,60,98,113,147)为大分子链转移剂,调控PS的聚合,在聚合过程中,都先形成聚合物微球,随着PS链段的增加,发生了从球到柱的转变。随着链段PDMAEMA聚合度的增加,得到的纳米线的直径也从15 nm增加到22,46,59,73 nm,最后增加到103 nm。
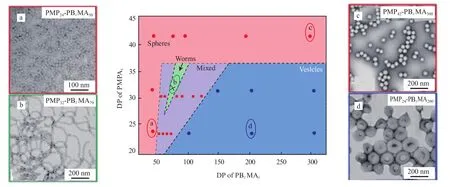
图1 PMPx-PBzMAy在甲醇中的PISA相图Fig.1 The polymerization-induced self-assembly(PISA) phase diagram constructed for PMPx-PBzMAy(diblock copolymer nano-objects prepared) in methanol. Conditions:64 ℃,20%(w) solids.
除了采用上述有机溶剂为聚合溶剂,水作为PISA的聚合溶剂具有更多聚合优势。水相聚合体系,是自由基聚合相对于离子聚合的优势,具有绿色无污染、价格低廉等特点,得到的聚合物微球可直接使用,无有机物排放问题。同时,在自由基聚合中,还具有链转移常数低、单体转换率高、聚合快等优点。然而在水相聚合中,可以选择用作B链段的单体较少,常见的单体包括,N-异丙基丙烯酰胺(NIPAM),N,N′-二乙基丙酰胺(DEAA),2-甲氧基丙烯酸乙酯(MEA),甲基丙烯酸2-羟丙酯(HPMA)以及甲基丙烯酸寡聚乙二醇甲基醚酯等(PEGMA)。
An等[44]第一个报道了水相中的PISA聚合,使用聚N,N′-二甲基丙烯酰胺(PDMA)为大分子链转移剂,微波引发,在70 ℃调控NIPAM的聚合,得到PDMA为壳,聚NIPAM为核的聚合物微球,表征结果显示,得到的聚合物微球尺寸在100~120 nm之间,其聚合物微球的固含量可达到15%(w)。使用类似的温敏性单体,加入双功能团单体作为交联剂,稳定核结构,就可得到具有温敏性能的纳米凝胶[45-46],如使用聚甲基丙烯酸寡聚乙二醇甲基醚酯为大分子链转移剂,调控丙烯酸与MEA聚合,使用低温引发剂在人体体温附近引发聚合得到了温敏纳米凝胶。
除了以上温度聚合物外,MEA和HPMA单体也被常常用于水相中的PISA聚合,HPMA单体在水中有着较大的溶解度,但是聚HPMA在水中不溶,因而可用做水相PISA。除了聚合物不溶于水外,HPMA单体还具有聚合速率快,转化率高,聚合物分布窄等特点。由于以上诸多的优点,以聚HPMA为B段,使用不同的A段,使用PISA方法得到了不同形貌的聚合物纳米微球。Armes等[47]报道了使用大分子链转移剂(PGMA47),在水中调控HPMA的聚合,发现了聚合诱导的蠕虫状胶束到囊泡的形貌转变。在该体系中,聚合可以明显的分为三个阶段,第一阶段(0~20 min)为诱导期;第二阶段(20~60 min)为溶解阶段,嵌段聚合物在溶液中表现为单分子分散状态;60 min之后进入第三阶段,形成了聚合物纳米微球。
3 PISA方法机理研究
Ji等[48]采用动态光散射和静态光散射相结合的技术,以聚环氧乙烯(PEO)大分子链为引发剂引发NIPAM聚合,研究PISA过程。按照光强和自组装尺寸把PISA过程分为三个阶段:诱导期阶段,单分子阶段和胶束化阶段。在第一阶段中,没有聚合的发生,聚合物链都是以单分子链的状态存在,光强较弱。第二阶段中,聚合开始,光强开始缓慢增加,这个阶段中第二嵌段长度还较小,大部分的聚合物链都仍然以单分子的形态存在,还有一小部分聚合物链形成松散的聚集体。随着聚合的进行,第二嵌段的长度增加,松散的聚集体数目增加,聚集数变大,光强增加,同时单分子的峰开始减弱。随着聚合的进一步进行,第二嵌段聚合度达到一个临界值,这一长度被定义为临界聚集聚合度(CMDP),这时嵌段共聚物在溶液中不能以单分子的状态存在,松散的聚集体也变的不稳定。嵌段共聚物倾向于聚集起来形成胶束,体系进入第三阶段。在第三阶段初期,在光散射上可看到单体峰和松散聚集体峰同时变小,而胶束的峰则出现随着时间延长而增加的现象。此时Rg/Rh(Rg为根均方半径,Rh为流体力学半径)大于1,表明胶束具有比较松散的结构。结合动力学数据可知,在同一时间内聚合速率受到较大的抑制,这是由于形成的胶束结构较紧密,限制了单体扩散的速率,因此聚合速率降低。随着聚合的进一步进行,聚合物第二嵌段的长度进一步增加,胶束变得更加紧密。Rg/Rh下降到0.6左右,表明了形成的胶束具有一个致密的结构。
在一个理想的使用活性PISA的过程中,所有的嵌段共聚物的分子链应有着相同的生长速度,链段的长度超过CMDP后,由于溶液中的嵌段共聚物在热力学上不稳定,导致开始形成聚集体。在形成胶束的初期,体系存在着单分子链与胶束的平衡,因此体系中有大量的单分子链。随着聚合的进行,疏溶剂链段的聚合度随着聚合的进行而增加。平衡向胶束方向移动,同时胶束的尺寸也增加。当所有的单分子都转变为胶束后,胶束重排使得胶束的结构更加致密,聚集数增加,因此光强的增加非常剧烈。
以上工作都是以稀溶液体系为主,利用溶剂的选择性作为自组装的驱动力制备纳米微球,单体浓度对溶剂的选择性影响不大。
在PISA过程中,溶剂对聚合物的溶解性、单体在核中的溶胀、链段的玻璃化转变温度两链段之间的不相容能力等都有着非常重大的影响[49]。例如,在第三阶段时,HPMA的聚合速度是第二阶段中的5倍。这是由于在形成聚合物微球之后,HPMA单体在PHPMA的核中溶胀,局部的单体浓度高,因而聚合速度加快,同时由于单体的溶胀,也有利于聚集体形貌的转变。
聚合链段之间的不相容性,由Flory-Huggins常数(χ)和聚合度的乘积决定。当聚合度相同的情况下,χ就决定了相分离能力,较高的χ会促进PISA过程的发生。例如引入离子基团,就可大大的增加χ,从而促进PISA中相分离的发生。Zhang等[50]以甲基丙烯酰氧乙基三甲基六氟化磷铵为模型化合物,以聚PEGMA和聚乙二醇(PEG)为大分子链转移剂调控聚合。研究发现在相同的聚合条件下,以聚PEGMA为A段的情况下,PISA得到的形貌发生从球状到蠕虫状到网状的变化,而在PEG为A段的情况下,则出现的是从球状到层状到网状的变化,这表明χ对PISA得到的聚集体形貌有着重要的影响。
4 结语
PISA方法是一个崭新的聚合物纳米微球的制备方法,将活性聚合与嵌段聚合物自组装结合起来,在活性聚合的过程中充分利用嵌段聚合物的自组装性质,得到各种结构的聚合物自组装体,不但可以用于稀溶液中,还可以用于制备高固含量的纳米微球,相对传统的聚合物自组装体制备方法,PISA方法更加简单且适合于大规模生产,制备得到的微观组装体可进一步修饰,并与无机材料负载制备催化剂等,是一种极具工业应用潜力的聚合方法。
工业上合成聚合物微球较多的方法是乳液聚合以及悬浮聚合,具有高效,高固含量,操作容易等特点。同PISA方法相比,使用乳液聚合合成100~1 000 nm范围的聚合物微球速度更快,固含量更好,分布更窄。但是合成20~50 nm范围的聚合物微球,PISA方法则优势明显,且PISA方法不需要加入表面活性剂调控微球尺寸,因而省略除去表面活性剂步骤,减少了操作费用。同时,由于PISA方法中,使用的是两嵌段制备得到的聚合物微球,增加溶解的A链段的长度,可减少聚合物微球之间的相互作用,使得到的聚合物微球结构更加稳定。此外使用PISA方法,更容易制备囊泡和柱状结构的聚合物。
使用CRP方法实现PISA,可用于水相聚合,同乳液聚合及悬浮聚合相比,可创造出更加丰富的结构和形貌。同时具有方法简单,可重复性好,后处理步骤简单(一步法)等特点。这种方法还可用于高浓度的聚合,一步法得到具有纳米尺度上自组装的凝胶,可具有复杂的相结构,用于电子、光电技术的应用。由于CRP方法是基于普通自由基聚合方法,工业放大相对简单,因而具有工业应用的潜力。
[1] Hamley I W. The physics of block copolymers[M].Oxford:Oxford Publish Press,1999:1-432.
[2] Bates F S,Fredrickson G H. Block copolymers-designer soft materials[J].Phys Today,1999,52(2):32-38.
[3] Kataoka K,Harada A,Nagasaki Y. Block copolymer micelles for drug delivery:Design,characterization and biological significance[J].Adv Drug Del Rev,2001,47(1):113-131.
[4] Bronstein L M,Chernyshov D M,Karlinsey R,et al. Mesoporous alumina and aluminosilica with Pd and Pt nanoparticles:Structure and catalytic properties[J].Chem Mater,2003,15(13):2623-2631.
[5] Zhang L F,Eisenberg A. Multiple morphologies of “crewcut”aggregates of polystyrene-b-poly(acrylic acid) blockcopolymers[J].Science,1995,268 (5218):1728-1731.
[6] Thurmond K B,Kowalewski A T,Wooley K L. Watersoluble knedel-like structures:The preparation of shell-crosslinked small particles[J].J Am Chem Soc,1996,118(30):7239-7240.
[7] Bridgestone Corporation. Nano-particle preparation and applications:US6437050[P].2002-8-20.
[8] Wang Xiaorong,Hall J E,Warren S Krom J,et al. Synthesis,characterization,and application of novel polymeric nanoparticles[J].Macromolecules,2007,40 (3):499-508.
[9] Charleux B,Delaittre G,Rieger J,et al. Polymerizationinduced self-assembly:From soluble macromolecules to block copolymer nano-objects in one step[J].Macromolecules,2012,45(17):6753-6765.
[10] Qu Qingwu,Liu Guangyao,Lü Xiaoqing,et al. In situ cross-linking of vesicles in polymerization-induced self-assembly[J].ACS Macro Lett,2016,5(3):316-320.
[11] Byard S,Williams M,McKenzie B E,et al. Preparation and cross-linking of all-acrylamide diblock copolymer nano-objects via polymerization-induced self-assembly in aqueous solution[J].Macromolecules,2017,50(4):1482-1493.
[12] Chen Shengli,Shi Pengfei,Zhang Wangqing. In situ synthesis of block copolymer nano-assemblies by polymerizationinduced self-assembly under heterogeneous condition[J].Chin J Polym Sci,2017,35 (4):455-479.
[13] Zhou Wei,Qu Qingwu,Xu Yuanyuan,et al. Aqueous polymerization-induced self-assembly for the synthesis of ketonefunctionalized nano-objects with low polydispersity[J].ACS Macro Lett,2015,4(5):495-499.
[14] Liu Guangyao,Qiu Qian,Shen Wenqing,et al. Aqueous dispersion polymerization of 2-methoxyethyl acrylate for the synthesis of biocompatible nanoparticles using a hydrophilic RAFT polymer and a redox initiator[J].Macromolecules,2011,44(13):5237-5245.
[15] Wu Juanjuan,Tian Chun,Zhang Lifen,et al. Synthesis of soap-free emulsion with high solid content by differential dripping RAFT polymerization-induced self-assembly[J].RSC Adv,2017,7:6559-6564.
[16] Otsu T,Yoshida M. Role of initiator-transfer agent-terminator(iniferter) in radical polymerization:Polymer design by organic disufides as iniferters[J].Makromol Chem Rapid Commun,1982,3 (1):127.
[17] Hawker C J. Molecular-weight control by a living free-radical polymerization process[J].J Am Chem Soc,1994,116(24):11185-11186.
[18] Hawker C J,Bosman A W,Harth E. New polymer synthesis by nitroxide mediated living radical polymerizations[J].Chem Rev,2001,101 (12):3661-3688.
[19] Kato M,Kamigaito M,Sawamoto M,et al. Polymerization of methyl-methacrylate with the carbon-tetrachloride dichlorotris(triphenylphosphine)ruthenium(Ⅱ) methylaluminum bis(2,6-di-tert-butylphenoxide) initiating system-possibility of living radical polymerization[J].Macromolecules,1995,28 (5):1721-1723.
[20] Chiefari J,ChongY K,Ercole F,et al. Living free-radical polymerization by reversible addition-fragmentation chain transfer:The RAFT process[J].Macromolecules,1998,31(16):5559-5562.
[21] Chiefari J,Mayadunne R T A,Moad C L,et al. Thiocarbonylthio compounds (S==C(Z)S—R) in free radical polymerization with reversible addition-fragmentation chain transfer (RAFT polymerization). Effect of the activating group Z[J].Macromolecules,2003,36 (7):2273-2283.
[22] Lowe A B,Mccormick C L. Reversible addition-fragmentation chain transfer (RAFT) radical polymerization and the synthesis of water-soluble (co)polymers under homogeneous conditions in organic and aqueous media[J].Prog Polym Sci,2007,32 (3):283-351.
[23] Destarac M,Brochon C,Catala J M,et al. Macromolecular design via the interchange of xanthates (MADIX):Polymerization of styrene with O-ethyl xanthates as controlling agents[J].Macromol Chem Phys,2002,203 (16):2281-2289.
[24] Destarac M,Bzducha W,Aton D,et al. Xanthates as chaintransfer agents in controlled radical polymerization (MADIX):Structural effect of the O-alkyl group[J].Macromol Rapid Commun,2002,23 (17):1049-1054.
[25] Ji Wenxi,Li Zichen,Chen Erqiang,et al. One-pot synthesis of nanoparticles based on styrene and maleic anhydride[C]// Fouth East Asian Polymer Conference. Tianjin:Chinese Chemical Society,2006:218-220.
[26] Zheng G H,Pan Caiyuan. Reversible addition-fragmentation transfer polymerization in nanosized micelles formed in situ[J].Macromolecules,2006,39 (1):95-102.
[27] Sugihara S,Sugihara K,Armes S P,et al. Synthesis of biomimetic poly(2-(methacryloyloxy)ethyl phosphorylcholine)nanolatexes via atom transfer radical dispersion polymerization in alcohol/water mixtures[J].Macromolecules,2010,43(15):6321-6329.
[28] Delaittre G,Nicolas J,Lefay C,et al. Surfactant-free synthesis of amphiphilic diblock copolymer nanoparticles via nitroxide-mediated emulsion polymerization[J].Chem Commun,2005,1(5):614-616.
[29] Blanazs A,Ryan A J,Armes S P. Predictive phase diagrams for RAFT aqueous dispersion polymerization:Effect of block copolymer composition,molecular weight,and copolymer concentration[J].Macromolecules,2012,45(12):5099-5107.
[30] Zhou Heng,Liu Chonggao,Gao Chengqiang,et al. Polymerization-induced self-assembly of block copolymer through dispersion RAFT polymerization in ionic liquid[J].J Polym Sci,Part A:Polym Chem,2016,54(11):1517-1525. .
[31] Han X,Poliakoff M. Continuous reactions in supercritical carbon dioxide:Problems,solutions and possible ways forward[J].Chem Soc Rev,2012,41(4):1428-1436.
[32] Semsarilar M,Jones E R,Blanazs A,et al. Efficient synthesis of sterically-stabilized nano-objects via RAFT dispersion polymerization of benzyl methacrylate in alcoholic media[J]. Adv Mater,2012,24(25):3378-3382.
[33] Rager T,Meyer W H,Wegner G,et al. Influence of chain length and salt concentration on block copolymer micellization[J].Macromolecules,1997,30(17):4911-4919.
[34] Liu Tianbo,Nace V M,Chu B. Cloud-point temperatures of BnEmBnand PnEmPntype triblock copolymers in aqueous solution[J].J Phys Chem B,1997,101 (41):8074-8078.
[35] Creutz S,van Stam J,de Schryver F C,et al. Dynamics of poly((dimethylamino)alkyl methacrylate-block-sodium methacrylate) micelles. Influence of hydrophobicity and molecular architecture on the exchange rate of copolymer molecules[J].Macromolecules,1998,31 (3):681-689.
[36] Brannan A K,Bates F S. ABCA tetrablock copolymer vesicles[J].Macromolecules,2004,37 (24):8816-8819.
[37] Yu Yisong,Zhang Lifeng,Eisenberg A. Morphogenic effect of solvent on crew-cut aggregates of apmphiphilic diblock copolymers[J].Macromolecules,1998,31(4):1144-1154.
[38] Pei Yiwen,Lowe A B. Polymerization-induced self-assembly:Ethanolic RAFT dispersion polymerization of 2-phenylethyl methacrylate[J].Polym Chem,2014,5(7):2342-2351
[39] Chen Shengli,Shi Pengfei,Zhang Wangqing. In situ synthesis of block copolymer nano-assemblies by polymerization induced self-assembly under heterogeneous condition[J].Chin J Polym Sci,2017,35(4):455-479.
[40] Warren N J,Armes S P J. Polymerization-induced selfassembly of block copolymer nano-objects via RAFT aqueous dispersion polymerization[J].J Am Chem Soc,2014,136: 10174-10185.
[41] Cai Weimin,Wan Wenming,Hong Chunyan,et al. Morphology transition in RAFT polymerization for formation[J]. Soft Matter,2010,6(21):5554-5561.
[42] Andreas H,Yang Pengcheng,Alexander N K,et al. Phosphonic acid-functionalized diblock copolymer nano-objects via polymerization-induced self-assembly:Synthesis,characterization,and occlusion into calcite crystals[J].Macromolecules,2016,49(1):192-204.
[43] Zhang Wenjian,Hong Chunyan,Pan CaiYuan. Fabrication and characterization of silica nanotubes with controlled dimensions[J].J Mater Chem,2014,2:7819.
[44] An Zesheng,Shi Qihui,Tang Wei,et al. Facile RAFT precipitation polymerization for the microwave-assisted synthesis of well-defined,double hydrophilic block copolymers and nanostructured hydrogels[J].J Am Chem Soc,2007,129(46):14493-14499.
[45] Grazon C,Rieger J,Sanson N,et al. Study of poly(N,N-diethylacrylamide) nanogel formation by aqueous dispersion polymerization of N,N-diethylacrylamide in the presence of poly(ethylene oxide)-b-poly(N,N-dimethylacrylamide) amphiphilic macromolecular RAFT agents[J].Soft Matter,2011,7(7):3482-3490.
[46] Qiu Qian,Liu Guangyao,An Zesheng. Efficient and versatile synthesis of star polymers in water and their use as emulsifiers[J].Chem Commun,2011,47(47):12685-12687.
[47] Armes S P,Battaglia G,Ryan A J. Mechanistic insights for block copolymer morphologies:How do worms form vesicles[J].J Am Chem Soc,2011,133(41):16581.
[48] Ji Wenxi,Yan Jingjing,Liang Dehai,et al. In situ and online monitoring polymerization-induced micellization[J]. Macromolecules,2008,41 (13):4914-4919.
[49] Cockram A A,Neal T J,Derry M J,et al. Effect of monomer solubility on the evolution of copolymer morphology during polymerization-induced self-assembly in aqueous solution[J]. Macromolecules,2017,50 (3):796-802.
[50] Zhang Baohua,Lü Xiaoqing,An Zesheng. Modular monomers with tunable solubility:Synthesis of highly incompatible block copolymer nano-objects via raft aqueous dispersion polymerization[J]. ACS Macro Lett,2017,6(3):224-228.
(编辑 杨天予)
Progress in the self-assembly of block copolymers of polymerizationinduced self-assembly
Sun Ruliu
(Sinopec Beijing Research Institute of Chemical Industry,Beijing 100013,China)
A new method of preparing block copolymer nanoparticles,the polymerization-induced selfassembly(PISA) method is described. The PISA method combines the concept of controlled radical polymerization and the self-assembly of block copolymer,which is recognized as an efficient route to produce block copolymer nanoparticles of controlled size,morphology,and surface chemistry. We discussed the advantages and drawbacks of PISA method,as well as the future research direction.
block copolymer;self-assembly;living polymerization
1000-8144(2017)07-0953-07
TQ 316.32
A
10.3969/j.issn.1000-8144.2017.07.020
2017-01-18;[修改稿日期]2017-04-27。
孙汝柳(1984—),女,山西省太谷县人,硕士,工程师,电话 010-59202538,电邮 sunruliu.bjhy@sinopec.com。
