γ石墨炔衍生物结构稳定性和电子结构的第一性原理研究∗
2017-08-09陈献程梅娟吴顺情朱梓忠
陈献 程梅娟 吴顺情 朱梓忠
(厦门大学物理系,半导体光电材料及其高效转换器件协同创新中心,厦门 361005)
γ石墨炔衍生物结构稳定性和电子结构的第一性原理研究∗
陈献 程梅娟 吴顺情 朱梓忠†
(厦门大学物理系,半导体光电材料及其高效转换器件协同创新中心,厦门 361005)
(2016年12月19日收到;2017年3月11日收到修改稿)
通过基于密度泛函理论的第一原理计算,系统研究了γ石墨炔衍生物的结构稳定性、原子构型和电子性质.γ石墨炔衍生物的结构是由碳六元环以及连接六元环间的碳链组成,碳链上的碳原子数为N=1—6.研究结果表明,碳链上碳原子数的奇偶性对γ石墨炔衍生物的结构稳定和相应的原子构型、电子结构性质具有很大的影响.其奇偶性规律为∶当六元环间的碳原子数为奇数时,体系中的碳链均为双键排布,系统呈现金属性;当六元环间的碳原子数为偶数时,系统中的碳链形式为单、三键交替排列,体系为直接带隙的半导体.直接带隙的存在能够促进光电能的高效转换,预示着石墨炔在光电子器件中的应用优势.N=2,4,6的带隙分布在0.94—0.84 eV之间,带隙的大小与碳链上三键的数量和长度有关.研究表明,将碳原子链引入到石墨烯碳六元环之间,通过控制引入的碳原子个数可以调控其金属和半导体电子特性,为设计和制备基于碳原子的可调控s-p杂化的二维材料和纳米电子器件提供了理论依据.
∶石墨炔,碳原子链,sp/sp2杂化,第一性原理计算
PACS∶71.20.—b,73.22.—f,31.15.Ar,71.15.MbDOI∶10.7498/aps.66.107102
1 引 言
碳是组成地球上所有已知生命的基础,多种杂化方式(sp,sp2,sp3)赋予了碳独特的性质,具有与几乎所有元素结合的能力.碳有众多的同素异形体,但曾经在很长的一段时间里,人们对碳同素异形体的认识停留在金刚石(sp3)和石墨(sp2)上.1985年,富勒烯(C60)[1]的发现彻底改变了这种情况.对碳成键的新认知打开了人们对碳的其他同素异形体合成的大门,各种新型的纳米碳材料被制备出来,如碳纳米管[2]、石墨烯[3,4]、石墨烯纳米带[5]、纳米环[6]、一维原子碳链(carbyne)[7−14]等.从本质上说,碳材料丰富的同素异构现象是sp,sp2,sp3不同的轨道杂化方式在碳材料的组成中所占的比例不同而造成的宏观上的差异.
由于英国曼切斯特大学的安德烈·海姆和康斯坦丁·诺沃肖洛夫等对石墨烯[15]的开创性研究引发了人们对石墨烯的强烈关注.石墨烯是由碳原子组成的,具有稳定的单层二维结构,其电子能带结构在费米能级(Fermi energy)呈现狄拉克锥特征,拥有优异的热导和电子迁移率;其热导率可达5000 W/mK,是金刚石的3倍[16],载流子迁移率达15000 cm2/Vs[17]是目前已知的具有最高迁移率的锑化铟材料的两倍,超过商用硅片迁移率的10倍以上.因此,石墨烯被广泛应用于电子器件,如在SiC表面成功合成的高性能外延石墨烯场效应晶体管,实现了240 nm栅极长度下100 GHz的截止频率[18];与5d系列过渡金属原子相结合的石墨烯显示了在自旋电子器件方面的应用潜力[19],以及石墨烯超胞在谷电子器件方向的应用[20].石墨烯的发现使得碳材料的研究进入了一个新的阶段,激发了科学家们对新型碳的同素异形体的研究热忱和兴趣.近年来,一种新的碳的同素异形体——石墨炔,因其独特的结构和优秀的电子、光学和机械性质[21,22],以及在纳米电子和储能[23]方面的可能应用,引起了人们的广泛关注.Baughman等[24]理论预测石墨炔是可以稳定存在的,它是由sp和sp2杂化混合组成的二维单层纳米碳材料.sp杂化碳原子键的存在打乱了石墨烯规则的六角晶格结构,从而形成具有不同结构和性质的石墨炔[24−33].2010年,Li等[30]利用六炔基苯在铜片表面上通过偶联反应成功合成了大面积的石墨炔(graphdiyne).随后他们又合成了石墨炔纳米管[30],这些实验的成功预示了石墨炔合成的可能性.一些关于石墨炔的理论研究也在进行,Kang等[22]通过第一性原理计算,研究了γ-graphyne的弹性、电子和光学性质.他们发现γ-graphyne比石墨烯更软,且拥有各向异性的光学性质.另外一些理论计算探究了γ-graphyne(graphdiyne)的储能能力[31−33].结果表明,它们可能具备的大容量储氢和储锂能力使得石墨炔在储能方面有着非凡的应用前景.Malko等[29]发现,α-,β-和6,6,12-graphyne有着类似石墨烯的电子结构,它们的能带在费米能级附近同样表现出了狄拉克锥特征.6,6,12-graphyne甚至表现出了比石墨烯更惊人的电子特性.除此之外,文献中也出现一些石墨炔潜在应用的研究,比如在锂离子电池电极材料[34,35]、气体分离[36]和海水淡化等[37]领域中的应用.
尽管石墨烯拥有独特的电学和机械性能,但带隙的缺失却让人们无法对石墨烯的导电性能进行调控.在纳米器件的设计和应用中,材料带隙的可调控性很重要,零带隙就限制了石墨烯在一些场效应晶体管和高速逻辑开关装置方面的应用[38−41].人们采取了一些方法来试图打开石墨烯的带隙,如氢化作用[42−47]、施加应力应变[48−51]等. 在石墨炔方面,拥有天然直接带隙的γ-graphyne似乎更适合在电子器件方面的应用[52].本文系统研究了γ-graphyne衍生物的原子构型和电子结构性质,重点关注了γ-graphyne衍生物的稳定性、原子构型以及能带结构与其六元环间碳原子数的关联规律.研究表明,碳链上碳原子数的奇偶性对γ石墨炔衍生物的结构稳定和电子结构性质具有很大的影响.将碳原子链引入到石墨烯碳六元环之间,通过控制引入的碳原子个数可以调控其金属和半导体电子特性,为设计和制备二维材料和纳米电子器件提供理论依据.
2 计算方法
本文的计算基于密度泛函理论.使用的第一性原理方法基于VASP程序包(Vienna ab initio simulation package)[53],该程序包采用平面波展开和映射缀加波势方法(projector augmented-wave potentials,PAW)[54].交换关联泛函采用Perdaw-Burke-Ernzerhof的广义梯度近似(GGA-PBE)[55],波函数的切断动能为500 eV.布里渊区的积分采用了Monkhorst-Pack网格方法,网格分辨率为2π×0.02−1.由于在z-方向上原胞的晶格常数很大,故在z方向仅采用了一个k点[56].为排除周期镜像原子的干扰,在垂直石墨炔平面的晶胞z方向上设置了厚度为20 Å的真空层.计算时,对所建立的模型的原胞形状和原胞内的原子位置都进行了充分的弛豫,原子弛豫时无预设对称性约束.总能的收敛、Hellman-Feynman力的收敛分别设置为10−4eV/atom,0.01 eV/Å.在获得最优化的几何结构的基础上计算系统的电子性质.
3 结果与讨论

图1 γ-N石墨炔材料的结构示意图(N=2)Fig.1.Atomic structures of γ-N(N=2).
本文的研究对象是γ石墨炔系列衍生物.图1给出的是一个γ石墨炔的典型结构,即γ-2结构(通式为γ-N结构,本文研究的是N=1—6结构,N为碳链上的原子数).γ-N结构可以看成是由碳六元环和连接六元环之间的碳链构成,其中碳链上的碳原子由N(这里N=1—6)个碳原子组成.从化学键角度看,γ-N结构可以看成是由不同比例的杂化和杂化碳原子组成∶位于碳六元环上的碳原子为,位于碳链上的碳原子为杂化.为考察碳六元环间的碳原子数N对γ-N结构稳定性及原子和电子结构的影响,本文对6个γ-N(N=1—6)结构进行了系统的几何优化及电子结构分析.
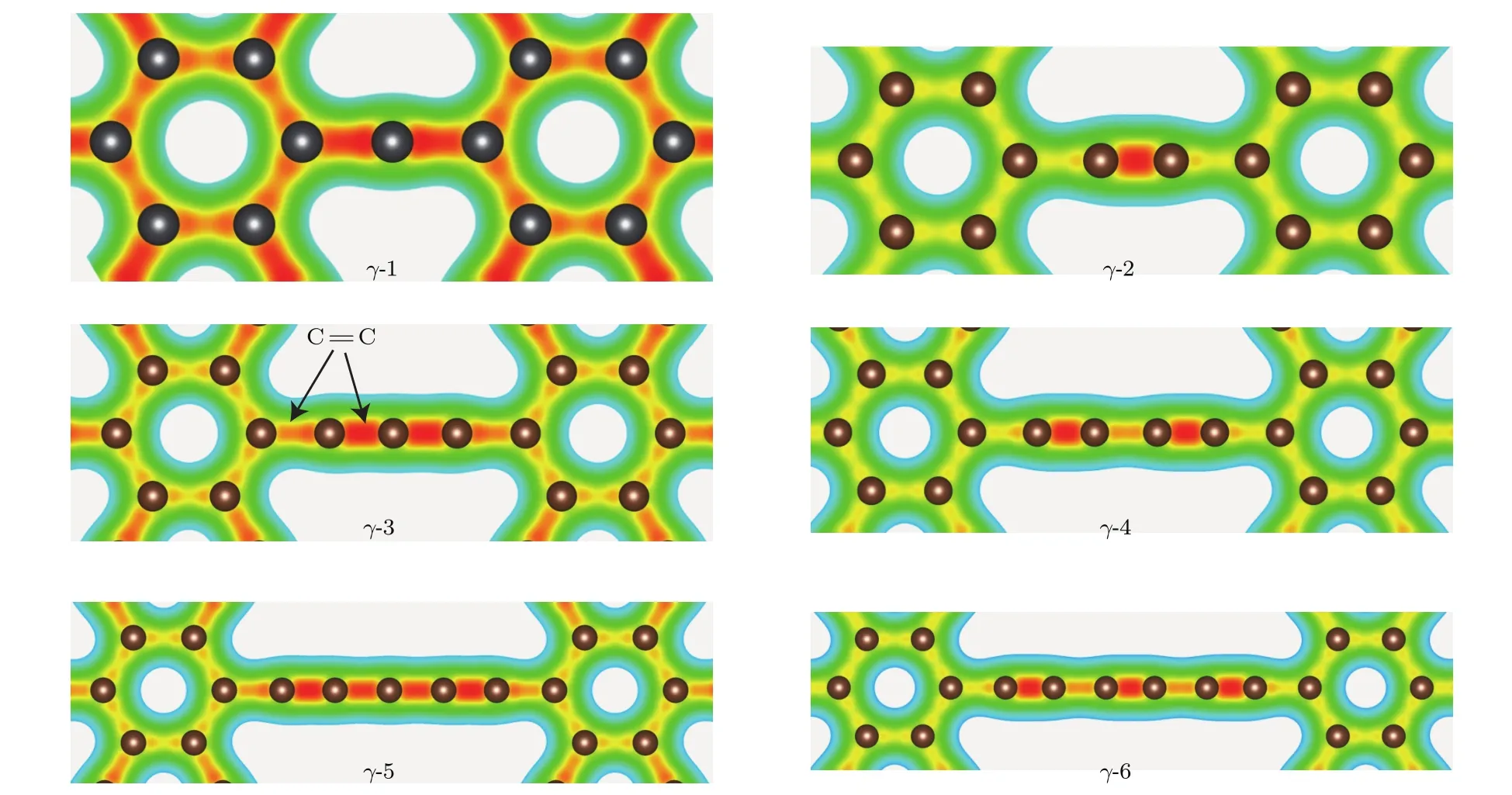
图2 (网刊彩色)γ-N石墨炔系列材料的电荷密度图Fig.2.(color online)Charge density of γ-N.
我们首先分析γ-N系列结构中的碳原子成键的情况.图2是γ-N石墨炔系列的电荷密度图,同一张图中颜色越深(越是红色)表示电荷密度越大.可以发现,γ-N系列结构的碳六元环和碳链上的碳原子成键情况与碳链上碳原子数的奇偶性密切相关,有如下规律∶当γ-N碳链上的碳原子数为奇数时(N=1,3,5),碳链上的碳原子均以(=C=C=)碳碳双键的形式成键;当六元环间的碳原子数为偶数时(N=2,4,6),碳链上的碳原子以单键和三键(-C≡C-)的形式成键.
计算中,原胞的体积(或晶格常数)和各原子的位置都经过充分的弛豫,所以可得到最优的碳链的长度.体系的结合能可以部分地体现体系的结构稳定性,因此我们计算了γ石墨炔系列衍生物的结合能Eb,计算公式为∶

其中Etotal为体系的总能量,EC为单个碳原子的能量,nC为原胞中碳原子的数目.
表1给出了优化的晶格常数和相应的结合能,同样可以发现碳链上碳原子数为偶数的结构N=(2,4,6)的结合能总是比碳原子数为奇数的结构N=(1,3,5)的结合能为大,这表明γ-2,4,6结构比γ-1,3,5的结构更为稳定.另一方面,γ-2,4,6的稳定性会随着炔连接单元(-C≡C-)数量的增加而下降.在前言中我们提到的γ-4(graphdiyne)石墨炔结构已由中科院化学研究所成功合成[30],所以从理论上说,完全有理由相信比γ-4更为稳定的γ-2(graphyne)也能够被成功合成.

表1 优化的晶格常数与结合能,晶格常数参见图1Table 1.Optimized lattice constants and binding energies,lattice constants are shown in Fig.1.
现在讨论γ石墨炔系列衍生物各个结构的键长分布情况.根据上面的分析可以发现,碳链中是否含有炔连接单元(即-C≡C-)会对体系的性质产生重要影响,因此我们把奇数个碳原子的碳链结构和偶数个碳原子的碳链结构分开讨论.图3是γ-1—6石墨炔系列材料碳链上C—C键的键长分布情况,图中碳链和横坐标上的数字表示键的序号,γ-N结构碳链上的C—C键长分布规律依奇偶性的不同呈现明显的差异.我们先讨论碳链上的碳原子数为奇数的情况∶可以发现,靠近六元环的两个C=C双键总是最长的,范围在1.37—1.38 Å,位于中央的C=C键长度相近,约为1.28 Å.靠近六元环的双键为C-sp2-C-sp杂化,与位于中央的C-sp-C-sp有所差异.当碳链上的碳原子数N为偶数时,C—C键长度呈长短交错分布.类似于奇数时的情况,靠近六元环的两个C—C单键是最长的,接近于1.40 Å,且不随着N的增大而变化.在中央的C≡C-C≡结构中,炔键的长度随着N的增大略微有所增长,但变化不大,范围在1.22—1.24 Å,两个炔键间的C—C键长接近于1.34.我们注意到,当碳原子链上的原子数为偶数时,链上单键的长度比传统的C—C单键(1.55)稍短,而三键的长度比传统的C≡C三键(1.20)稍长.因此,两个三键中的单键有类似双键的特征.图4中纵坐标的起点处表示石墨烯的C—C键长,可以看到,γ-N石墨炔系列结构的六元环的碳碳键长稍大于石墨烯的键长,且六元环上的sp2C—C键长的变化同样存在着奇偶性差异.原子链的原子数为奇数时,六元环中C—C键长分布在1.44—1.45 Å,随着N的增大键长逐渐减小.当原子链的原子数为偶数时,六元环中的碳碳键长分布在1.42—1.43 Å,随着N的增大,键长有些许波动.

图3 γ-1—6碳链上C—C键的键长分布情况 (a)奇数链系列;(b)偶数链系列Fig.3.Bond length of C—C on carbon chain,for(a)odd and(b)even carbon chains.

图4 γ-N体系六元环上的C—C键长随碳链长度的变化Fig.4.The length change of C—C on hexagonal rings of γ-N.

图5 γ-N(N=1—6)石墨炔衍生物的平均结合能随碳原子链上碳原子数N的关系Fig.5.The relationship between the binding energy and the number of carbon atom on the chain of γ-N(N=1–6).

图6 (网刊彩色)γ-N石墨炔系列材料(N=1—6)的能带结构与态密度图.倒易空间高对称点分别为K(−1/3,2/3,0),Γ(0,0,0),M(0,1/2,0)Fig.6.(color online)Band structure and density of states of γ-N.
我们知道,六元环和碳链上的碳原子是sp2和sp杂化的,γ-N石墨炔系列衍生物可以看成是两种杂化按一定的比例混合组成的.而石墨烯和一维碳链可以分别看成是γ-N系列石墨炔材料的两个极端,即全部由sp2杂化组成的石墨烯和全部由sp杂化组成的一维碳链.为了理解γ-N石墨炔材料的能量稳定性,我们计算了各种γ-N(N=1—6)石墨炔结构的结合能(一个碳平均的结合能),并且与石墨烯的结果做对比(同样平均到一个碳上),结果如图5所示,图中横坐标表示碳链上的原子数N.从图5可以看出,单个碳平均的结合能和碳链上的碳原子数的奇偶性有关∶当碳链上的原子数为奇数时,平均单个碳原子的结合能随N的增加而减小;当碳链上的碳原子数为偶数时,平均碳结合能随N的增加而增大;而且,碳链上原子数为偶数的平均结合能总是小于碳链上原子数为奇数的情况.这表明∶双键连续排列的结构的稳定性会随着碳链上碳原子数N的增加而增大;单、三键交替排列的结构的稳定性则随着石墨炔网络中炔连接单元(-C≡C-)数量的增加而下降.同时,单、三键交替排列的结构总是比双键连续排列的结构要稳定,即-C(sp)≡C(sp-和=C(sp)=C(sp)=结构虽然都是sp杂化,但前者比后者更稳定.所以,碳链上的碳原子更愿意形成单、三键交替的(-C≡C-)结构,这与由前面计算出的体系的结合能所得出的结论一致.
为了分析γ-N石墨炔系列衍生物的电子结构性质,我们计算了这些材料的能带结构和态密度(DOS),计算结果如图6所示.从图中可以看到,原子链中原子数为偶数(N为偶数)的系列材料与原子链中原子数为奇数(N为奇数)的系列材料的能带结构和态密度与原子链上原子数N的奇偶性存在明显的依存特征.计算结果显示,六元环间的碳原子数N为奇数的γ-N系列材料呈现金属性特征,而N为偶数的γ-N系列材料则呈现半导体特征.γ-2材料的带隙出现在布里渊区的M点,带隙值为0.941 eV,γ-4材料的带隙出现在布里渊区的Γ点,带隙值为0.894 eV,γ-6材料的带隙又出现在M点,带隙值为0.842 eV.可见带隙的数值随着炔键数量的增加而减小,且带隙的位置出现在M点或Γ点与γ-N炔键数量的奇偶性有关.同时我们还发现,带隙的大小还与炔键的长度密切相关.
γ-2石墨炔材料的炔键长度的理论计算值为1.22 Å,带隙0.941 eV,晶格常数6.89 Å.我们固定住γ-2材料的原胞的晶格常数,并改变炔键d的长度,发现炔键长度d与带隙之间的关系基本上呈线性相关,参见图7(b).随着d的增大,带隙逐渐减小.当d≥1.29 Å时,带隙消失,电子结构出现狄拉克锥.这些特征表明,γ石墨炔中三键对带隙的影响是Peierls畸变在二维体系中的表现,与打开石墨烯的带隙相类似[57,58].

图7 (a)γ-2石墨炔中炔键长度d;(b)γ-2材料的带隙与炔键长度d的关系;(c)当γ-2石墨炔的炔键d=1.287 Å时的能带结构Fig.7.(a)Definition of the triple bond length d of γ-2;(b)energy gap in γ-2 as a function of d;(c)electronic band structure of γ-2 for d=1.287 Å.
4 结 论
本文通过第一原理方法系统地计算了γ石墨炔衍生物的结构稳定性、相应的原子构型和电子结构性质,发现六元环间的sp杂化碳原子所形成的碳链是影响γ石墨炔衍生物的原子构型和电子结构的主要因素.能带结构与石墨炔中原子链上的原子数之间有明显的奇偶性规律∶当六元环间的碳原子数N为奇数时,γ-N系列材料的碳链形成连续的双键排布,体系呈现金属性;当六元环间的碳原子数N为偶数时,γ-N系列结构的碳链形成单、三键交替排列,体系为直接带隙的半导体.带隙的大小与碳链上三键的数量和长度有关.碳链上具有偶数碳原子的γ-N结构比具有奇数的γ-N结构更为稳定.本研究对设计和制备基于碳原子的可控sp,sp2杂化混合的二维材料及纳米电子器件具有参考价值.
[1]Kroto H W,Heath J R,O’Brien S C,Curl R F,Smalley R E 1985 Nature 318 162
[2]Iijima S 1991 Nature 354 56
[3]Novoselov K S,Geim A K,Morozov S V,Jiang D,Zhang Y,Dubonos S V,Grigorieva I V,Firsov A A 2004 Science 306 666
[4]Castro Neto A H,Guinea F,Peres N M R,Novoselov K S,Geim A K 2009 Rev.Mod.Phys.81 109
[5]Li X,Wang X,Zhang L,Lee S,Dai H 2008 Science 319 1229
[6]Kong X Y,Ding Y,Yang R,Wang Z L 2004 Science 303 1348
[7]Chuvilin A,Meyer J C,Algara-Siller G,Kaiser U 2009 New J.Phys.11 083019
[8]Jin C H,Lan H P,Peng L M,Suenaga K,Iijima S 2009 Phys.Rev.Lett.102 205501
[9]Fan X F,Liu L,Lin J Y,Shen Z X,Kuo J L 2009 ACS Nano 3 3788
[10]Liu M J,Artyukhov V I,Lee H,XufB,Yakobson B I 2013 ACS Nano 7 10075
[11]Liu Y,Jones R O,Zhao X L,Ando Y 2003 Phys.Rev.B 68 125413
[12]Zhao X L,Ando Y,Liu Y,Jinno M,Suzuki T 2003 Phys.Rev.Lett.90 187401
[13]Cao R G,Wang Y,Lin Z Z,Ming C,Zhuang J,Ning X J 2010 Acta Phys.Sin.59 6438(in Chinese)[曹荣根,王音,林正喆,明辰,庄军,宁西京2010物理学报59 6438]
[14]Qiu M,Zhang Z H,Deng X Q 2010 Acta Phys.Sin.59 4162(in Chinese)[邱明,张振华,邓小清 2010物理学报59 4162]
[15]Novoselov K S,Geim A K,Morozov S V,Jiang D,Zhang Y,Dubonos S V,Grigorieva I V,Firsov A A 2004 Science 306 666
[16]Balandin A A,Ghosh S,Bao W Z,Calizo I,Teweldebrhan D,Miao F,Lau C N 2008 Nano Lett.8 902
[17]Chen J H,Jang C,Xiao S D,Ishigami M,Fuhrer M S 2008 Nanotechnology 3 206
[18]Lin Y M,Dimitrakopoulos C,Jenkins K A,Farmer D B,Chiu H Y,Grill A,Avouris P 2010 Science 327 662
[19]Sun M L,Tang W C,Ren Q Q,Zhao Y M,Du Y H,Yu J,Du Y H,Hao Y T 2016 Physica E 80 142
[20]Wang S K,Wang J 2015 Phys.Rev.B 92 075419
[21]Narita N,Nagai S,Suzuki S,Nakao K 1998 Phys.Rev.B 58 11009
[22]Kang J,Li J,Wu F,Li S S,Xia J B 2011 J.Phys.Chem.C 115 20466
[23]Srinivasu K,Ghosh S K 2012 J.Phys.Chem.C 116 5951
[24]Baughman R H,Eckhardt H,Kertesz M 1987 J.Chem.Phys.87 6687
[25]Coluci V R,Braga S F,Legoas S B,Galvao D S,Baughman R H 2004 Nanotechnology 15 S142
[26]Falcao E H L,Wudlf2007 J.Chem.Technol.Biotechnol.82 524
[27]Hirsch A 2010 Nat.Mater.9 868
[28]Enyashin A N,Ivanovskii A L 2011 Phys.Status Solidi B 248 1879
[29]Malko D,Neiss C,Vines F,Gorling A 2012 Phys.Rev.Lett.108 086804
[30]Li G X,Li Y L,Liu H B,Guo Y B,Li Y J,Zhu D B 2010 Chem.Commun.46 3256
[31]Zhang H,Zhao M,He X,Wang Z,Zhang X,Liu X 2011 J.Phys.Chem.C 115 8845
[32]Srinivasu K,Ghosh S K 2012 J.Phys.Chem.C 116 5951
[33]Li C,Li J,Wu F,Li S S,Xia J B,Wang L W 2011 J.Phys.Chem.C 115 23221
[34]Jang B,Koo J,Park M,Lee H,Nam J,Kwon Y,Lee H 2013 Appl.Phys.Lett.103 263904
[35]Hwang H J,Koo J,Park M,Park N,Kwon Y,Lee H 2013 J.Phys.Chem.C 117 6919
[36]Zhao W H,Yuan L F,Yang J L 2012 Chin.J.Chem.Phys.25 434
[37]Lin S C,Buehler M 2013 J.Nanoscale 5 11801
[38]Novoselov K S,Jiang D,Schedin F,et al.2005 Proc.Natl.Acad.Sci.USA 102 10451
[39]Ma Y,Dai Y,Guo M,Huang B 2012 Phys.Rev.B 85 235448
[40]Brumfel G 2009 Nature 458 390
[41]Kaloni T P,Cheng Y C,Schwingenschloegl U 2012 J.Mater.Chem.22 919
[42]Elias D C,Nair R R,Mohiuddin T M,et al.2009 Science 323 610
[43]Singh A K,Yakobson B I 2009 Nano Lett.9 1540
[44]Balog R,Jorgensen B,Nilsson L,et al.2010 Nat.Mater.9 315
[45]Ma Y,Dai Y,Guo M,Niu C,Zhang Z,Huang B 2012 Phys.Chem.Chem.Phys.14 3651
[46]Burgess J S,Matis B R,Robinson J T,et al.2011 Carbon 49 4420
[47]Castellanos-Gomez A,Wojtaszek M,Arramel,Tombros N,van Wees B J 2012 Small 8 1607
[48]Cocco G,Cadelano E,Colombo L 2010 Phys.Rev.B 81 241412
[49]Gui G,Li J,Zhong J 2008 Phys.Rev.B 78 075435
[50]Pereira V M,Castro Neto A H,Peres N M R 2009 Phys.Rev.B 80 045401
[51]Ni Z H,Yu T,Lu Y H,Wang Y Y,Feng Y P,Shen Z X 2008 ACS Nano 2 2301
[52]Schirber M 2012 Physics 5 24
[53]Kresse G,Furthmuller J 1996 Comput.Mater.Sci.6 15
[54]Blochl P E 1994 Phys.Rev.B 50 17953
[55]Perdew J P,Burke K,Ernzerhof M 1996 Phys.Rev.Lett.77 3865
[56]Monkhorst H J,Pack J D 1976 Phys.Rev.B 13 5188
[57]Lee S H,Chung H J,Heo J,Yang H,Shin J,Chung U I,Seo S 2011 ACS Nano 5 2964
[58]Peierls R E 1955 Quantum Theory of Solids(Clarendon:Oxford)p108
PACS∶71.20.—b,73.22.—f,31.15.Ar,71.15.MbDOI∶10.7498/aps.66.107102
*Project supported by the National Key Research and Development Program(Grant Nos.2016YFA0202601,2016YFB0901502).
†Corresponding author.E-mail:zzhu@xmu.edu.cn
First-principle study of structure stability and electronic structures of γ graphyne derivatives∗
Chen Xian Cheng Mei-Juan Wu Shun-Qing Zhu Zi-Zhong†
(Department of Physics,Semiconductor Optoelectronic Material and High Efficiency Conversion Device Collaborative Innovation Center,Xiamen University,Xiamen 361005,China)
19 December 2016;revised manuscript
11 March 2017)
A new carbon allotrope—graphyne has attracted a lot of attention in the field of material sciences and condensedmatter physics due to its unique structure and excellent electronic,optical and mechanical properties.First-principles calculations based on the density functional theory(DFT)are performed to investigate the structures,energetic stabilities and electronic structures of γ-graphyne derivatives(γ-N).The studied γ-graphyne derivative consists of hexagon carbon rings connected by onedimensional carbon chains with various numbers of carbon atoms(N=1–6)on the chain.The calculation results show that the parity of number of carbon atoms on the carbon chains has a great influence on the structural configuration,the structural stability and the electronic property of the system.The γ-graphyne derivatives with odd-numbered carbon chains possess continuous C—C double bonds,energetically less stable than those with evennumbered carbon chains which have alternating single and triple C—C bonds.The electronic structure calculations indicate that γ-graphyne derivatives can be either metallic(when N is odd)or direct band gap semiconducting(when N is even).The existence of direct band gap can promote the efficient conversion of photoelectric energy,which indicates the advantage of γ-graphyne in the optoelectronic device.The band gaps of γ-2,4,6 are between 0.94 eV and 0.84 eV,the gap decreases with the number of triple C—C bonds increasing,and increases with the augment of length of carbon chains in γ-2,4,6.Ourfirst-principles studies show that introducing carbon chains between the hexagon carbon rings of graphene gives us a method to switch between metallic and semiconducting electronic structures by tuning the number of carbon atoms on the chains and provides a theoretical basis for designing and preparing the tunable s-p hybridized two-dimensional materials and nanoelectronic devices based on carbon atoms.
∶graphyne,carbon chain,sp/sp2hybrid,first-principle calculations
∗国家重点研发计划(批准号:2016YFA0202601,2016YFB0901502)资助的课题.
†通信作者.E-mail:zzhu@xmu.edu.cn
©2017中国物理学会Chinese Physical Society
